Antibody-dependent cell-mediated cytotoxicity (ADCC) is a potent mechanism of cytotoxicity, mainly mediated by natural killer (NK) cells. When specific antibodies bind to the targeted cell membrane, immune cells induce cell death through this response. This is one of several major mechanisms by which antibodies limit and control infection.
The interaction of antibodies and immune cells occurs through a family of receptors: Fc receptors (FcR). In humans, the FcR family of IgG (FcγR) consists of six receptors: FcγRI/CD64, FcγRIIa/CD32a, FcγRIIb/CD32b, FcγRIIc/CD32c, FcγRIIIa/CD16a, and FcγRIIIb/CD16b, of which CD16a is mainly responsible for triggering NK cell mediation Guided ADCC. Therefore, understanding the mechanism of action of CD16a can help us to maximize the potential of NK cells by improving monoclonal antibodies, thereby expanding the scope of cancer immunotherapy.
The mechanism of action of ADCC
NK cells are natural lymphocytes that are very effective at destroying stressed cells, such as virus-infected or tumor-transformed cells. Normally, the ADCC effect mainly activates NK cells through FCR-binding antibodies. The most characteristic FcR on the NK cell membrane is FcγRIII (CD16), in which NK cells mainly express CD16a, while CD16b is limited to neutrophils. Human NK cells are divided into two major subpopulations: CD56bright and CD56dim. CD56brightNK cells are potent cytokine producers but lack CD16a; whereas CD56dim are highly cytotoxic and express CD16a.
When IgG-opsonized targets are recognized by CD16a, NK cells release different cytotoxic molecules, which trigger the death of target cells. This mechanism relies on the formation of immune synapses and the degranulation of lysed granules containing perforin and granzymes. In addition to degranulation, NK cells can clear target cells by binding target death receptors such as DR4, DR5 or Fas to their death receptor ligands such as FasL and TRAIL.
The biology of CD16a
CD16a is a transmembrane receptor with a short C-ter cytoplasmic tail and two extracellular Ig-like domains. Its intracellular part does not have any signaling components, therefore, in order to transmit signals, it requires two signaling chains with immunoreceptor tyrosine-based activation motifs (ITAMs).
CD16a interacts with CH2 of IgG in a 1:1 manner, in which the N-glycan chain at N297 on CH2 also plays a key role in this interaction. Therefore, not only the amino acid sequence, but also its glycan composition can greatly influence the affinity of CD16a for Fc and thus the potency of ADCC.
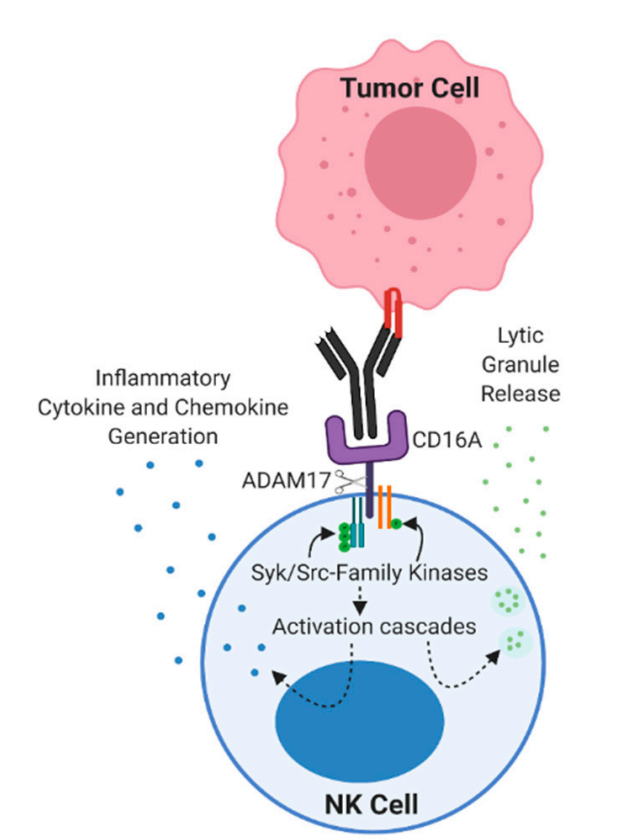
CD16a does not have any ITAM domains in its cytoplasmic tail and thus requires the help of two ITAM domain-containing intracellular chains in tandem, CD3ζ and FcϵRIγ. Upon CD16a binding, the kinase Lck belonging to the Src family is activated and phosphorylates the ITAM domains of CD3ζ and/or FcϵRIγ. Phosphorylated ITAM allows the recruitment and phosphorylation of kinases from the Syk family, such as Syk and ZAP-70, which in turn are responsible for subsequent signaling. Among their substrates, PI3K is highly related because it converts PIP2 to PIP3, which releases IP3 and DAG upon PLC-γ treatment. DAG activates the PKC family, which helps trigger degranulation. IP3 induces calcium release from the endoplasmic reticulum, and this calcium influx, one of the major signals triggered by ADCC, also leads to the translocation of NFAT in the nucleus, inducing transcription of its target genes. Other pathways involved in activation by CD16a also contribute to ADCC, such as the ERK2 MAPK pathway.
CD16a is rapidly downregulated after CD16a-dependent NK cell killing. One of the mechanisms of CD16a shedding is mediated by disintegrin and metalloproteinase 17 (ADAM17), which is expressed on NK cells. Cleavage of CD16a by ADAM17 occurs in the cis structure, which means that NK cells expressing ADAM17 cannot induce CD16a shedding on another NK cell. Another mechanism of CD16a downregulation upon activation is internalization, which occurs not only on CD16a but also on other intracellular signaling components such as CD3ζ, ZAP-70, and Syk.
Strategies to improve the effect of ADCC
cytokine
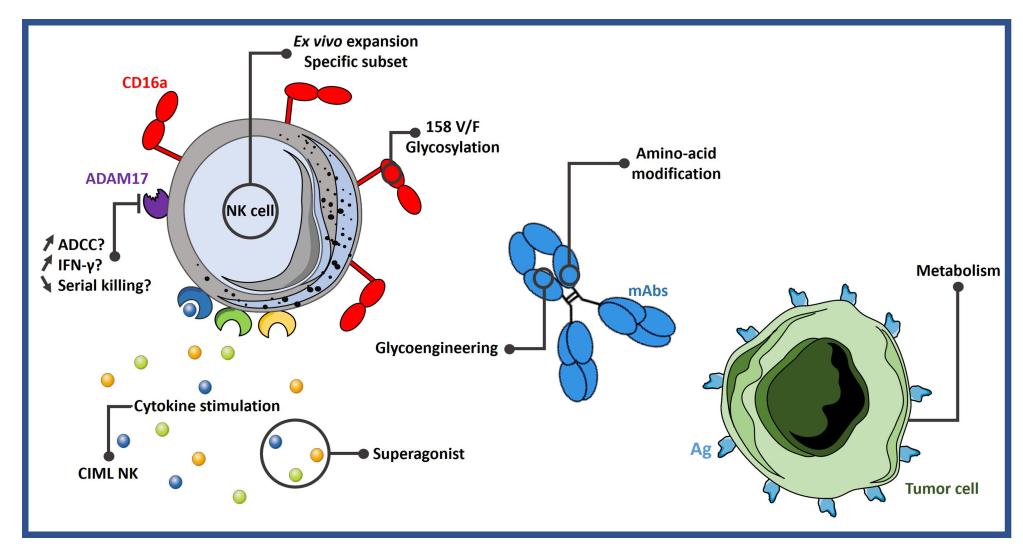
A simple way to increase ADCC activity is to stimulate NK cells with proinflammatory cytokines. NK cell transplantation has been successfully performed in several clinical trials, such as induction of memory-like (CIML) NK cells by infusion of cytokines, the most commonly used combinations of cytokines include IL-12, IL-15, and IL-18. CIML NK cells express more IFN-γ than conventional NK cells and show superior cytotoxicity against leukemia cells and primary acute myeloid leukemia (AML) blasts.
IL-12 increases the production of IFN-γ and TNF-α by NK cells. In a phase I/II clinical trial, cetuximab was combined with IL-12 in unresectable or recurrent head and neck squamous cells cancer patients. Compared with patients with progression-free survival (PFS) less than 100 days, patients with PFS greater than 100 days had higher ADCC activity in vitro.
IL-15 is a cytokine necessary for the survival and function of NK and CD8+ T cells. N-803 is a mutant N72D IL-15 superagonist that has shown enhanced NK cell activity in vitro and in vivo in preclinical models and clinical trials. In addition, it increased the number of NK and CD8+ T cells.
Inhibit ADAM17
ADAM17 is the main driver of CD16a downregulation upon activation. One strategy to improve ADCC in NK cells is to prevent CD16a shedding by targeting ADAM17. The study found that using CRISPR/Cas9 technology to knock down ADAM17 in NK cells showed better IFN-γ production and ADCC activity in vitro and in vivo. However, it has also been shown that blocking ADAM17 reduces NK cell survival as well as CD16a-mediated serial killing, the inability of cells to separate from target cells, hindering their movement to another target cell.
In conclusion, the benefit of ADAM17 inhibition is unclear and the results are controversial. The success of this strategy awaits the results of an ongoing clinical trial (NCT04023071) evaluating iPSC-derived NK cells harboring non-cleavable CD16a in AML and B-cell lymphoma.
Engineered Antibodies
The Fc portion of the antibody is engineered to increase the affinity of the Fc for CD16a and subsequently improve ADCC. Some mutations are very prevalent, for example, the so-called GASDALIE, consisting of 4 substitutions in the Fc: G236A/S239D/A330L/I332E. This mutation showed a significant increase in affinity for CD16a with little increase in affinity for CD32b.
Another mutation, called mutant 18 (F243L/R292P/Y300L/V305I/P396L), significantly increased ADCC. These mutations have now entered the clinic as the anti-HER2 antibody margetuximab and have shown promising results in Phase I treatment of various HER2-positive cancers.
In addition, glycoengineering of antibody Fc N-glycans is expected to increase the affinity for CD16a. The most studied sugar modification is deglycosylation, such as defucose modification. This modification resulted in increased binding to CD16a and improved ADCC. This technology has been used with the anti-CD20 antibody obinutuzumab.
NK cell adaptor
In addition to engineering antibodies, the form of antibodies can also be studied. Based on the success of the bispecific T cell engager (BiTE) strategy, a similar format was developed for NK cells, the so-called bispecific killer cell engager (BiKE). BiKE consists of two scFvs via linker sequences, one single-chain antibody targeting CD16a and the other targeting the tumor antigen.
In addition, there is the trispecific killer cell engager (TRiKE), with 2 tumor antigens targeted together with CD16a, or the addition of IL-15 in place of a third single-chain antibody. The use of this structure allows NK cells to relocate to tumor cells and leads to a robust ADCC effect on target cells.
AFM13 is a bispecific antibody against CD16a and CD30, currently in Phase II clinical trials for the treatment of peripheral T-cell lymphoma, transformed mycosis fungoides and Hodgkin lymphoma. In an Ib clinical study for the treatment of relapsed or refractory Hodgkin lymphoma, the combination of AFM13 and pembrolizumab produced an objective response rate of 88% and an overall response rate of 83%. GTB-3550 is a CD16/CD33/IL-15 form of TRiKE that is currently being studied in a phase I/II trial in several types of leukemia (NCT03214666).
Regulate metabolic pathways
Metabolic dysregulation is a key driver of immunosuppression in the tumor microenvironment, and numerous studies have shown that immune cell metabolism is critical for its function. Recently, some metabolic drugs have shown promising results in enhancing NK cell activity. Metformin, has been shown to increase the expression of immune cell ligands, especially ICAM-1. ICAM-1 is an integrin that regulates intracellular signaling in lymphocytes, including NK cells. The use of metformin in combination with UCB to expand NK cells showed higher tumor cell clearance in vitro and in vivo.
Furthermore, another strategy is to directly modulate NK cell metabolism. cMyc is a key factor regulating the mechanisms of glycolysis and oxonate metabolism in mouse NK cells, and lack of cMyc results in impaired NK cell responses. Glycogen synthase kinase-3 (GSK3) mediates cMyc degradation in mouse NK cells. Consistent with this, GSK3 overexpression was detected in NK cells of AML patients and was associated with impaired cytotoxicity against AML cells, and GSK3 inhibitors restored the activity of these NK cells. Several clinical trials are currently evaluating the efficacy of the GSK3 inhibitor LY2090314 in solid and hematological cancers (NCT01632306, NCT01287520, and NCT01214603).
issues to be resolved
Targeting CD16a may help address some of the existing limitations of current cancer immunotherapy therapeutic antibodies, thereby maximizing the potential of ADCC to promote more favorable clinical outcomes. However, there are still some unanswered questions that need to be answered:
What are the advantages of targeting CD16a over other NK cell activating receptors?
What is the most efficient way to activate NK cells to trigger ADCC? To what extent can activated NK cells trigger adaptive immunity to enhance overall antitumor activity?
Can NK cell adaptors promote homing and persistence of NK cells at tumor sites?
What is the expression level of CD16a+ NK cells and macrophages in the tumor? Since CD16a is expressed on both NK cells and macrophages, it is necessary to understand the contribution of different immune cell types relative to different tumor entities when CD16a is specifically targeted.
What is the effect of CD16a shedding on ADCC activity and its clinical efficacy? Are there advantages or disadvantages to using non-shedding CD16-engineered cell products?
Does Downregulation of NK Cell Receptors or Targeted Tumor Antigens After First-Line Therapy Affect the Efficacy of NK Cell Engager Therapy?
How is the clinical efficacy of NK cell activators affected by parameters such as patient age, health status at the start of treatment, and conditioning regimen?
summary
Natural killer cells are a unique group of antitumor effector cells with functions such as MHC-independent cytotoxicity, cytokine production, and immune memory, making them key players in the innate and adaptive immune response systems. ADCC via CD16a is one of the main mechanisms by which NK cells exert effector functions. Therefore, an in-depth understanding of the biology of CD16a helps us to find strategies to enhance the effect of ADCC, thereby enhancing the antitumor activity of NK cells.
references:
1. From CD16a Biology to Antibody-Dependent Cell-Mediated Cytotoxicity Improvement. Front Immunol. 2022;13:913215.
2. Reimaginingantibody-dependent cellular cytotoxicity in cancer: the potential of naturalkiller cell engagers. Trends Immunol. 2022 Nov;43(11):932-946