The conjugation of Linker-drug (LD) to antibodies is a critical step in the generation of ADCs and the determination of key quality attributes of ADCs that can fundamentally alter the pharmacokinetics and therapeutic index of ADCs. More than 95% of ADCs currently are based on two conjugation techniques: cysteine alkylation or lysine acylation. Seven of the top ten approved ADCs are based on cysteine conjugation (Adcetris, Polivy, Padcev, Enhertu, Trodelvy, Blenrep, and ZynLonta); three are prepared based on lysine conjugation (Kadcyla, Mylotarg, and Besponsa) . In addition to the traditional two methods, new technologies are now emerging rapidly, not only involving innovative methods of cysteine (alkylation and crosslinking) and lysine (acylation or alkylation) coupling to Produces more homogeneous ADCs and improves therapeutic index, but also extends to new coupling techniques such as carbonyl, amide, azide based coupling techniques.
Sulfhydryl (-SH) Sulfhydryl (-SH) is one of the most prevalent reactions in the ADC field and is primarily based on three elements.
(A) Sulfhydryl groups are the most nucleophilic amino acid side chain functional groups (more nucleophilic than amines);
(B) The number of interchain cysteines suitable for binding in the antibody is relatively small (8 for IgG1 and IgG4) and is not critical for antibody stability, which enables relatively controllable DAR;
(C) Interchain cysteines, present in the hinge region, link the heavy (HC) and light (LC) chains and can ‘hide’ the hydrophobic payload after ligation.
Currently, thiol-coupled ADCs are basically based on maleimide chemistry (Figure 1). A variety of maleimide-based reagents have been developed for alkylation, which occurs on native interchain cysteines or engineered cysteines.
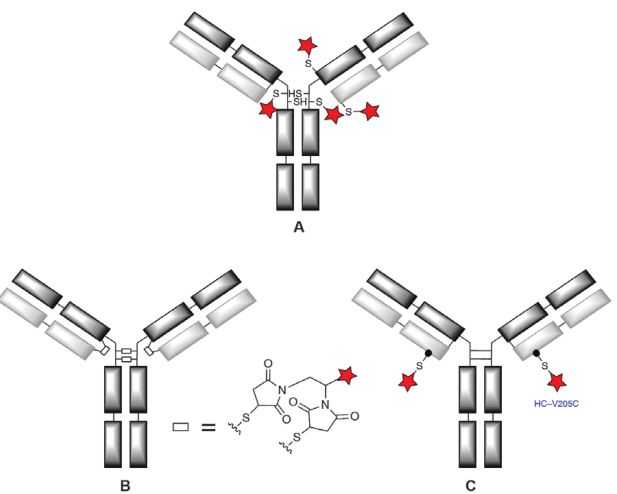
Figure 1. Groups based on maleimide chemistry to couple ADC thiols
Alkylation of Maleimide
Maleimide, the structural element for the reaction of maleic anhydride with amine derivatives, is easily alkylated by nucleophilic Michael addition to a thiol group to form a stable thioether bond (Figure 2). Maleimides react with thiols in the pH range of 6.5-7.5 at 1000 times the reaction rate of amines, allowing complete conversion with minimal reagents. The major disadvantage of maleimide alkylation is the reversibility of the Michael addition, the rate of which is highly dependent on the pKa of the specific cysteine residue to which it is attached. This inverse Michael reaction, which may lead to premature release of linker drug (LD) in the circulation and possibly adverse events, has also been an important driver for the development of more stable maleimide variants.
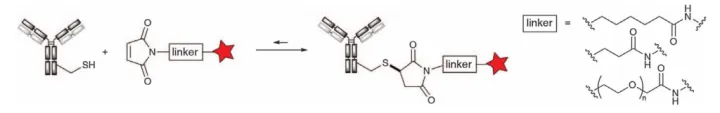
Figure 2. Cysteine Alkylation with Various Maleimide Reagents
Cysteines in mAbs are all involved in the formation of disulfide bonds, so a reduction step is required to release the thiol group before binding to LD. By optimizing the amount of reducing agent (usually TCEP or DTT) to release specific free thiol groups (up to 8 for IgG1 and IgG4). The marketed ADCs prepared by conjugation with cysteine are Adcetris, Polivy, Padcev, Enhertu, Blenrep, Trodelvy, Zynlonta, RC48, etc. Due to the lack of site specificity, these ADCs are often generated as random mixtures with average DARs between 2.3-4. However, with two exceptions, Enhertu and Trodelvy were generated by near-quantitative levels of binding of all interchain cysteines with DARs of 7.7 and 7.6, respectively, thus being site-specific. Although the maleimide-hexyl linker is used in the vast majority.
However, there are some ADCs with electron-deficient aminoethyl or ethylene glycol spacers that may inhibit the retro-Michael reaction, such as β-aminoethyl based AbGn-107, Zynlonta, ADCT-502, IMGN632, JBH492, MEDI2228 and TR1801-ADC ; Glycol-based SYD985, MORAb-202 and ZW49. Some phenyl-substituted maleimides, which are also reported to be more stable, are still in preclinical development. Subsequent investigators developed ADCs that bind LD to antibody-specifically designed cysteines at a site-specific basis, which may provide a higher therapeutic index than random ADC mixtures. Currently, several site-specific ADCs have entered the clinic, such as SGN-CD19B, CD123A and CD352A (all HC-S239C mutations); RG7861/DSTA4637S (LC-V205C mutations); IMGN632 (S442C mutations); BAT8003 (A114C) mutation); and ADCs based on double cysteine mutations, such as PF-06804103 (LC-K183C HC-K290C). Two other techniques have also been developed, namely cysteine insertions, such as MEDI2228 (HC-i239C); and HC-terminal polypeptide fusions, such as ALT-P7 (C-terminal ACGHAACGHA fusion). However, the use of cysteine-engineered antibodies does not guarantee clinical success and some have been abandoned, for example, BAT8003 and the three ADCs from Seagen mentioned above.
The undesired retro-Michael reaction can be inhibited by hydrolysis of the thioether-succinimide adduct to provide the maleic acid derivative. This hydrolysis can be achieved by prolonged treatment of ADCs at pH 9; however, this may lead to protein degradation events such as deamidation or formation of pyroglutamate. Seagen solved this problem by using DPR technology with an aminomethyl group in the a-position of the alkyl chain (Figure 3). After addition of the thiol, the amino group induces spontaneous hydrolysis of the succinimide conjugate, preventing the retro-Michael reaction. The ADC products that introduced this technology are SGN-CD48A (now discontinued), SGN-CD228A.
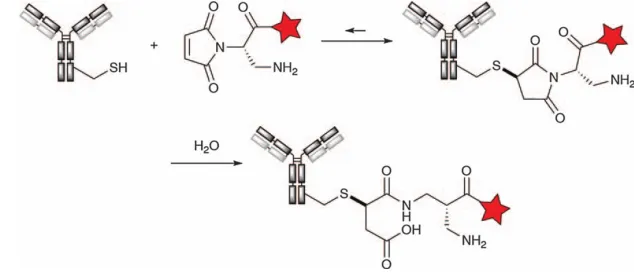
Figure 3. Alkylation of cysteine with diaminopropionic acid (DPR) aminomethyl-based maleimide reagent followed by autohydrolysis to maleic acid conjugates
Other Cysteine Alkylation Reagents
Various alternatives to maleimide alkylation have also been developed in recent years. For example, the high reactivity of alpha-haloacetamide reagents is well known, with 2-iodoacetamide being the undisputed reagent of choice for free cysteine capping. More importantly, the thioether bond formed by it is completely irreversible, so it is superior to thiol-maleimide ether in terms of stability.
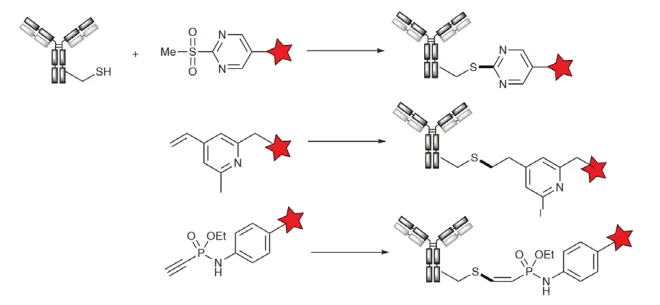
Additionally, Alley’s study using the α-bromoacetamidopropyl linker provided thioether ADCs with excellent plasma stability, with no measurable systemic drug release within 2 weeks of injection in mice. Since methanesulfonylbenzothiazole (MSBT) is a selective thiol blocking agent, Barbaset et al. developed a series of aromatic methanesulfonic acid reagents for the alkylation of thiol proteins. Similarly, mesylate pyrimidines react with thiols through a nucleophilic aromatic substitution mechanism (Fig. 4), a concept applied to SKB264. Researchers at Glythera (now Iksuda) report highly stable thioether ADCs obtained by reacting free cysteine thiol groups with 4-vinylpyridine, a technique called PermaLinks (Figure 4).
The resulting ADC has proven stability. The phosphine phosphite technology developed at the University of Berlin, now commercialized by Tubulis, shows that aryl phosphonimide compounds with electron-deficient triple bonds show an excellent cysteine compared to traditional maleimides. The amino acid selects the reactivity, while the resulting (Z)-vinyl sulfide has excellent stability (Figure 4).
Figure 4. Alternative techniques for cysteine alkylation
Bismaleimide or Bismaleamic Acid Alkylation/Crosslinking
The ability to alkylate maleimide has also been applied to various cross-linking bismaleimide reagents (Figure 5). Each reagent will capture two free cysteine thiol groups, resulting in a DAR4 ADC upon complete reduction of all eight interchain disulfide bonds. For example, the bismaleimide crosslinker is used in New Bio’s NBT828.
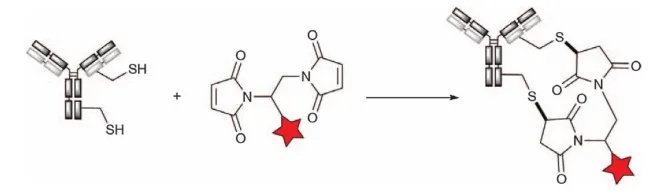
Figure 5. Bismaleimide Reagent Crosslinking Cysteine
Researchers from AstraZeneca report on engineering antibodies with bismaleimide-modified payload (PBD) heavy-bridged cysteine to obtain DAR1 ADCs (Figure 6).
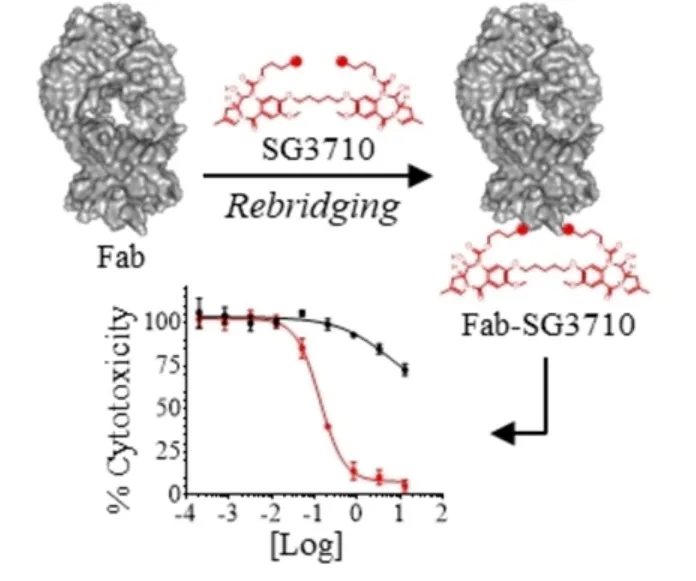
Figure 6. Bismaleimide-modified payload heavy-bridged cysteine engineered antibody
Alkylation/crosslinking of β-sulfone
The high nucleophilicity and facile Michael addition of free thiols have also been used in different types of crosslinkers, originally reported by Del Rosario and Wilbur and by Poly-Therics (now Abzena; THIObridge Technologies) for ADC development. Heavy bridges can increase the stability of the reduced antibody by relinking the formed thiol using a dithiol-reactive group, thereby better preserving the conformational integrity in the final linker. Treatment of the reduced antibody with the α-tosylmethyl-α,β-unsaturated ketone reagent formed in situ from the bis-a,a-tosylmethyl ketone precursor results in a series of reactions, e.g., Michael addition , β-elimination of tolyl sulfinic acid, and Michael addition again to provide a stable cross-linked product (Fig. 7). THIObridget technology is currently used in the OBI999 ADC (phase 1).
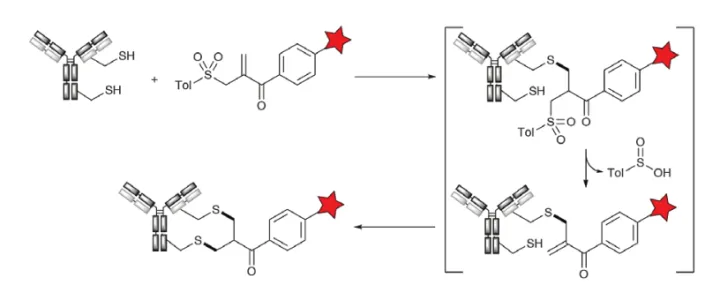
Figure 7. Crosslinking of Cysteine with Tosylate Acrylate Reagent (THIObridge Technology)
Alkylation/Crosslinking of Bisbromomethyl Reagents
A third method of cross-linking DAR4 ADCs was developed by Concortis (now Sorrento) with the C-lock technology. The C-lock technology combines the nucleophilicity of thiols with the nucleophilic SN2 substitution of brominated benzyl groups. Thus, thioethers are prepared by irreversible crosslinking by reduction of interchain disulfides followed by reaction with bisbromomethylaryl reagents, such as bisbromomethylquinoxaline (Figure 8). An ADC (CD38-ADC) using C-lock technology has entered the clinic.
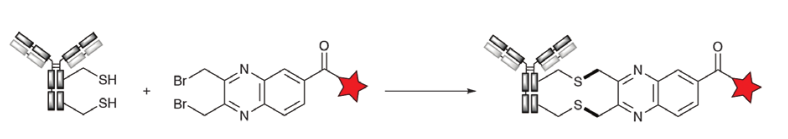
Figure 8. Dialkylated crosslinking (C-lock) of cysteine with bisbromomethylquinoxaline.
Alkylation/Crosslinking of Dibromoazepinediones
Inspired by the re-bridging of reductive disulfide bonds by dibromomaleimide groups, Chudasamaa et al. developed a re-bridging technique based on the core structure of azadione (PD) groups. Unlike maleimide derivatives, the azadione ring is more stable and does not undergo ring opening by hydrolysis, resulting in a more uniform conjugated compound. The researchers developed derivatives of monobromo PD (MBPD) and dibromo PD (DBPD) and demonstrated that both derivatives were able to react specifically with thiol groups at pH 8.0. The technique of heavy bridge chemistry using DBPD thiols has been used to create ADCs with highly controlled payload modifications (Figure 9).
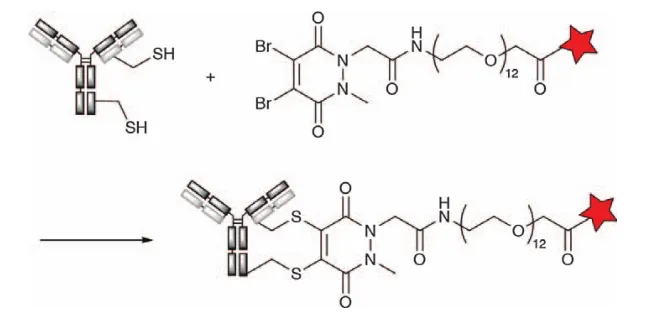
Figure 9. Cross-linking of Antibody Thiol Groups to Dibromoazadione Derivatives
Formation of disulfide bonds
Although not directly attached to antibody cysteines, disulfide bonds were used as part of linkers in early ADC development, such as MLN2704, CMD-401, BIWI-1, and IMGN242. A linker design containing a disulfide bond between the cytotoxic payload and the antibody can facilitate a reversible linkage that can be cleaved by glutathione reduction within tumor cells to release the toxin. However, premature cleavage of disulfide bonds in the circulation frequently occurs due to the presence of dissolved cysteines, which results in the release of the payload prior to entry into tumor cells. Adding protecting groups to carbon atoms near the disulfides creates steric hindrance to increase cycling stability, but in many cases they also reduce the rate of toxin release from tumor cells.
Furthermore, even with the increased stability of hindered disulfides, ADCs are not as efficient as drug binding at engineered cysteine residues at optimal positions in the antibody sequence. Pillowet et al. were the first to directly attach small-molecule drugs to engineered cysteines at different positions in the antibody (Figure 10). By comparing ADCs using hindered or unhindered disulfide bonds, it was determined that the engineered cysteine at the LC-K149C position was the most stable binding site for binding disulfide-containing drugs. ADCs created using the LC-K149C site exhibited excellent cycling stability while allowing efficient cleavage of glutathione within tumor cells.
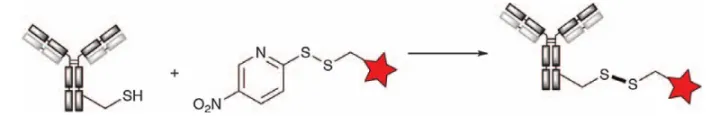
Figure 10. Disulfide bond generation by engineering cysteines to prepare antibody conjugates
Amino (–NH2)
Amino groups are naturally present in the lysine side chain and are the most convenient functional group for conjugation to mAbs, which do not require prior chemical modification steps. Although most of the protein is protonated at neutral pH due to its high pKa (~10.5), there is still a small fraction of amino groups in the protein that will exist in a neutral, unprotonated form. The amino group is second only to the thiol group in nucleophilicity, but clearly ahead of the side chains of other amino acids (eg, arginine, serine, tryptophan, methionine). Furthermore, the amino groups of lysines do not require reductive release like cysteine thiols and are available in large quantities, eg, antibodies may contain up to 80 solvent-exposed lysine residues.
The primary coupling reactions for amine compound modification go through two main routes: acylation or alkylation. In general, these reactions are fast and selective, produce stable bonds (amide or secondary amine bonds), and result in high yields. However, selective acylation of a limited number of lysines is very challenging due to the abundance of lysines, and thus lysine-conjugated ADCs typically show a broad DAR distribution with binding occurring at a large number of sites (Fig. 11).
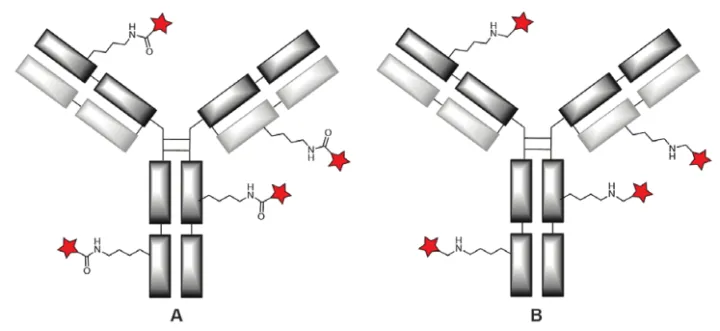
Figure 11. Random coupling to native lysine via acylation (A) or reductive alkylation (B)
Random acylation
Hydroxysuccinimide (NHS) esters are the most commonly used reactive derivatives of carboxylic acids for aminoacylation reactions. NHS ester-containing reagents are easily formed by the reaction of carboxylates with NHS in the presence of carbodiimide, and with the release of the NHS leaving group, they react with amine nucleophiles to form stable amide bonds (Figure 12). Competitive reactions of this ester with other nucleophiles, such as alcohols or water, are orders of magnitude slower, while reactions with thiols or histidine nitrogen groups yield unstable thioesters or imidazolyl esters that are easily hydrolyzed or remain Exchanges with adjacent amines to form amide bonds. In addition to neutral NHS esters, sulfonated derivatives (sulfo-NHS) with better water solubility can also be prepared. For example, the SMCC reagent used in the manufacture of Kadcylas.
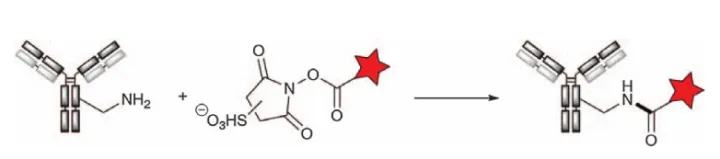
Figure 12. Acylation of amino groups with NHS esters
Representative ADCs prepared by lysine acylation are Mylotarg, Besponsa, IMGN853 and SAR408701. Wagneret et al. from the University of Strasbourg, France, report a new method via the reaction of 4-azidobenzoyl fluoride (ABF) with the amino group of lysine. This reagent reacts rapidly with primary amines on antibodies to form acylated products, which are more efficient than using NHS ester-based reagents (Figure 13). Its terminal azide group can be used to couple toxins, fluorescent dyes and oligonucleotides.
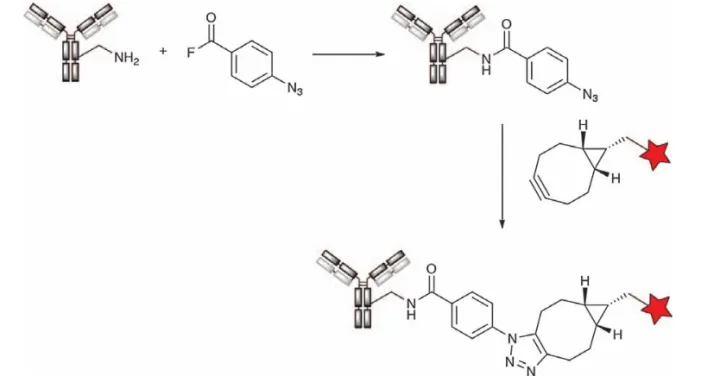
Figure 13. Reaction of 4-azidobenzoyl fluoride with lysine side chains to generate azide antibodies
Site-specific acylation
Although direct acylation of lysine side chains is the most straightforward and easiest method for antibody conjugation, the broad DAR distribution is a significant disadvantage. Therefore, site-specific acylation techniques were developed. For example, Concortis Biosystems (now Sorrento Therapeutics) reported a one-step lysine conjugation technology called K-lock, which can generate site-specific ADC. Although the details of the technology have not been disclosed, it has been used in various projects, such as a 5T4-targeting ADC drug (ZV05) in collaboration with Lova Therapeutics. Bernardes et al. also reported site-specific lysine acylation. Acylation reactions at slightly basic pH would be kinetically favorable using reagents from computer-aided predictions of antibody lysines with the lowest pKA. It was found that the sulfonyl acrylate reacted rapidly using only a single molar equivalent (37 °C, pH 8.0), enabling quantitative and irreversible modification of the trastuzumab light chain Lys207 with complete preservation of native secondary structure and function (Fig. 14).
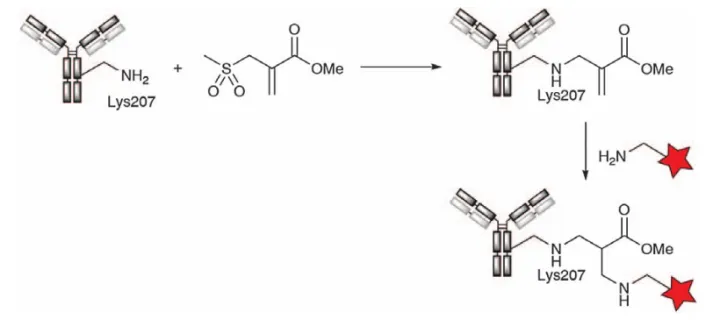
Figure 14. Reaction of Lys207 with methyl sulfonate followed by aza-Michael addition of amino-functionalized payloads
Researchers at Ajinomoto report a two-stage, site-directed lysine-conjugated Ajicap technology. At the heart of this technology are carefully designed high-affinity cyclic peptides from protein A that have high affinity to the Fc portion of antibodies. It was found that the acylation of Lys248 near the antibody-polypeptide complex was extremely selective if the cyclic peptide had a reactive ester moiety. Subsequent reduction of the disulfide moiety simultaneously removes the affinity peptide from the antibody and releases a free thiol group for reaction with the payload (Figure 15).
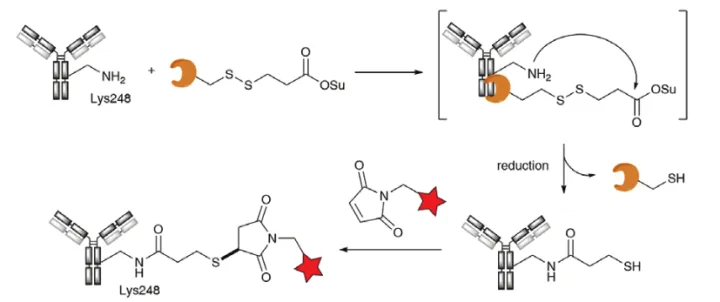
Figure 15. Ajicap technology based on antibody Fc binding to high affinity peptides
Interconversion of functional groups (NH2 to SH)
BMS’s BMS-986183 and BMS-936561 (MDX-1203) elegantly combine amino and thiol chemistries. Treatment of the antibody with Traut’s reagent, a five-membered iminothioester, resulted in the formation of the amidine (amidine) group on the lysine, ring opening and exposure of the free thiol (Figure 16). After removal of excess reagents, the desired ADC was readily obtained by reacting with a maleimide-functionalized linker. Clearly, ADCs prepared in this way, while interesting, may suffer from both the broad distribution of lysine coupling and the potential retro-Michael reaction of thiol groups.
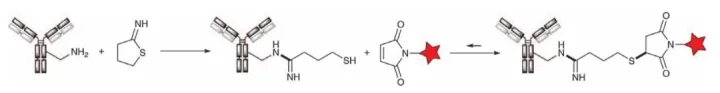
Figure 16. Antibody reacts with Traut reagent to introduce free thiol group for coupling via maleimide-based linker HTI-1066/SHR-A1403 (Hengrui) employs a Alternative to alcohol. The c-Met antibody was first treated with S-(3-oxopropyl)thioacetate to install a free thiol group on the native lysine. The payload was then introduced via maleimide conjugation (Figure 17).
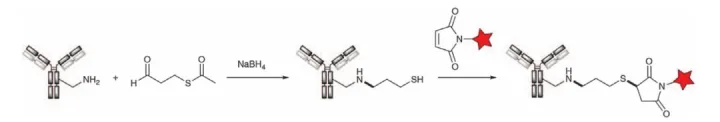
Figure 17. Lysine alkylation of S-(3-oxopropyl)thioacetate with antibodies to introduce free thiol groups and introduction of payloads via maleimide conjugation
Functional group interconversion (NH2 to maleimide)
One of the most common and successfully applied functional group interconversion methods is the bifunctional active ester/maleimide reagent SMCC or its sulfonated variant (sulfonated SMCC) to process native lysines. Thiol-functionalized linker-payloads were then introduced by Michael addition (Figure 18). Alternatively, the payload itself can be introduced if properly functionalized with a thiol moiety, as is the case for DM1, for example, which has been applied to several ADCs, most notably Kadcyla (T-DM1), DEBIO-1562, and Avid100.
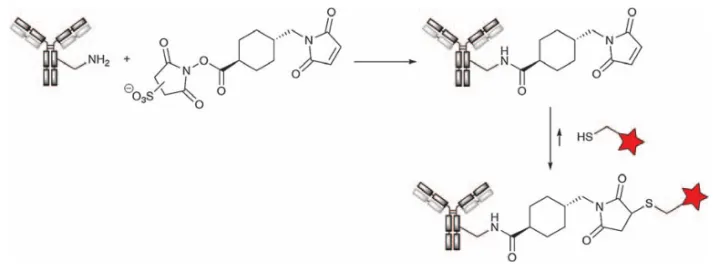
Figure 18. First treatment of native antibody with SMCC reagent followed by addition of thiol-functionalized payload to prepare ADC
references
1. Chemical Linkers in Antibody–Drug Conjugates (ADCs).2.Characterization of disulfide bond rebridged Fab–drug conjugates prepared using a dual maleimide pyrrolobenzodiazepine cytotoxic payload.3.Site-specific conjugation of native antibodies using engineered microbial transglutaminases.4.Structure and dynamics of a site-specific labeled Fc fragment with altered effector functions.
2. BioValley