ADC Drug Assessment Background
Before designing a DDI assessment strategy for a drug, an understanding of the properties of the drug itself is required. Therefore, we first review the relevant characteristics of Antibody-Drug Conjugates (ADC). ADC is composed of antibody + linker + toxic small molecule. Among them, antibodies ensure targeting, small molecules exert drug efficacy (such as anti-tumor cytotoxicity), and linkers ensure the stability of ADC in systemic circulation and degradability in target tissue cells. At present, the administration of ADC drugs is mostly injection, and most of the indications are anti-tumor.
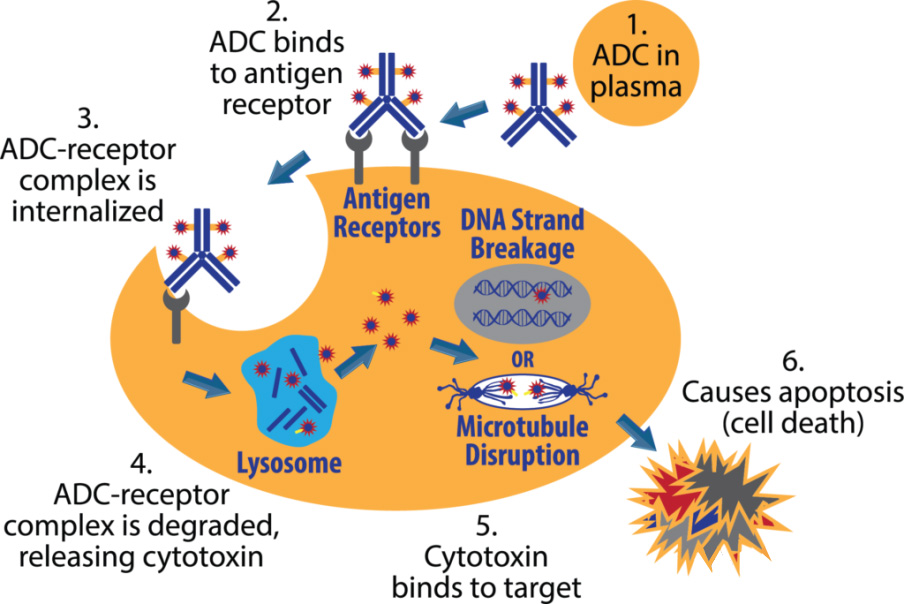
The mechanism of action of the ADC can be summarized as the following process:
After the ADC in the blood circulation binds to the target antigen on the surface of the tumor cell, it enters the cell through endocytosis;
Internalized antigen-ADC complexes enter the lysosomal pathway;
The acidic environment and proteases cause the linker to break and release toxic small molecules;
The released toxic small molecules act on DNA or microtubules in tumor cells, inhibit tumor DNA replication, and induce apoptosis.
The mechanism of action of ADC and the compositional characteristics of antibodies + small molecules also form a special metabolic clearance pathway in the body, including:
Target-mediated clearance TMDD (antibody signature)
Immunogenicity-mediated clearance (antibody signature)
Non-specific hydrolysis/metabolism (antibody characteristics)
Small molecule specific shedding of toxins (targeted shedding in target cells, antibody signature)
Metabolic clearance of free toxin small molecules (mediated by CYP metabolic enzymes and transporters)
It can be seen that the DDI-related metabolic enzyme transporter substrate research mainly focuses on toxic small molecules, while the antibody linker part is not eliminated by conventional drug-metabolizing enzyme transporter metabolism, so there is no need to conduct related substrate research.
DDI Assessment Guidelines
At present, the relevant DDI assessment perspectives and strategies for ADC drugs can refer to the following three guiding principles (white papers):
FDA “Drug-Drug Interaction Assessment for Therapeutic Proteins Guidance for Industry”
FDA “Clinical Pharmacology Considerations for Antibody-Drug Conjugates”
White Paper (industry consensus) “Current Approaches for ADME Characterization of Antibody-Drug Conjugates: An Industry White Paper”
The experience is summarized as follows:
1) The antibody part is performed according to the macromolecular DDI guidelines:
1.1) Antibodies related to the regulation of cytokines (immune levels);
1.2) In addition, it is also necessary to examine the DDI under the drug combination in the following three situations:
a) Combination with the same pharmacodynamic target drug;
b) Combination with drugs that interfere with the binding of ADC Fc region to IgG and FcRn;
c) If ADC PK is affected by immunogenicity, DDI between immunosuppressant and ADC needs to be considered.
2) Toxin small molecules are performed in accordance with guidelines based on metabolic enzymes and transporter DDI;
3) The main body of drug efficacy is small toxin molecules, and the safety window of small toxin molecules is narrow, so focus on small toxic molecules:
3.1) The exposure of toxin small molecules in the peripheral blood circulation is very low, and the risk of being a “perpetrator” (inhibitor inducer research) is extremely small;
3.2) Focus on the risk of toxic small molecules as “victims” (substrate research):
a) The effect of enzyme inducers on toxin small molecules: if the periphery does not play a major pharmacodynamic role, clinical DDI is not considered;
b) Effects of enzyme inhibitors on toxin small molecules: Increased peripheral concentrations can raise safety concerns and clinical DDI needs to be considered.
3.3) The DDI risk of toxin small molecule active metabolites needs to be investigated.
DDI Assessment Strategy
1) In vitro DDI assessment time:
Before IND application (indications are mostly anti-tumor, patients with tumors are directly enrolled in phase I, and there are concomitant drugs in tumor patients, it is necessary to provide scientific basis for concomitant drugs and risk control measures);
Clinical DDI can be directly incorporated into other phase I PK trials concurrently (see the case below).
2) In vitro DDI assessment strategy:
Listed small molecule toxins should directly refer to the application materials, and if necessary, supplement according to the requirements of the new guidelines or new indications;
New toxic small molecules complete a full set of assessment (in descending order of importance) according to the DDI guidelines based on metabolic enzymes and transporters:
a) Enzyme substrates (metabolic stability, identification of metabolic pathways)
b) Transporter substrates
c) Enzyme Inhibition Enzyme Induction
d) Transporter inhibition
e) CASE BY CASE principle (eg, targeting liver cancer cells, liver enzyme inhibition transporter inhibition studies need to be considered)
Case analysis
Based on the published application materials and literature, the DDI research conducted during the development of Adcetris is summarized as follows:
1) Introduction to the drug:
Trade name: Adcetris
Generic name: brentuximab vedotin (Bentuximab for injection)
ADC drug composed of monoclonal antibody targeting CD30 + MMAE
Treatment of adult patients with relapsed or refractory systemic anaplastic large cell lymphoma (sALCL) or CD30-positive Hodgkin lymphoma (HL)
2) In vitro DDI research content and results:
2.1) MMAE is a P-gp substrate, but has no strong inhibitory effect on P-gp;
The transmittance of B→A at 1, 10 and 100uM is much higher than that of A→B. The efflux ratio was significantly decreased in the presence of P-gp inhibitors;
Slightly inhibits the activity of P-gp on transporting Digoxin, with IC50 greater than 50uM.
2.2) MMAE is not a substrate of BCRP, MRP2, OATP1B1, OATP1B3, OCT2, OAT1 and OAT3;
2.3) MMAE is a substrate of CYP3A4;
2.4) The metabolites of MMAE in human hepatocytes were detected in both rat and monkey hepatocytes.
2.5) C4, C7, and C8 are the main metabolites of MMAE:
Metabolite in vitro activity assessment showed that C4, C7 and C8 were cytotoxic to CD30 positive tumor cells with IC50 of 0.9nM, 31nM and 0.01nM, respectively (=MMAE);
Metabolite generation pathway identification found that C4 and C8 were generated by CYP3A4, and C7 was generated by CYP3A4 and CYP2D6.
2.6) MMAE has no induction effect on CYP1A2, 2B6, 2C8, 2C9, 2C19 and 3A;
2.7) MMAE has no reversible inhibitory effect on CYP1A3, 2B6, 2C8, 2C9, 2C19, 2D6 and CYP3A metabolizing testosterone, but has inhibitory effect on CYP3A metabolizing midazolam activity, IC50=10uM;
2.8) MMAE has a time-dependent effect on CYP3A, the effect is derived from the intermediate product C10, kinact and KI are 0.10min-1 and 1.12uM, respectively
3) Clinical DDI research content
3.1) Trial Title: Assessment of DDI between Adcetris and CYP3A4 Substrate Drugs, Inhibitors and Inducers and Assessment of Their Excretion Routes
3.2) The main purpose of the test:
a) Drug Interactions:
PK Effect of Adcetris on CYP3A4 Substrate Drug Midazolam PK Effect of CYP3A4 Inhibitor Ketoconazole on Adcetris PK Effect of CYP3A4 Inducer Rifampicin on Adcetris
b) Identify the main route of excretion of MMAE
3.3) The secondary purpose of the test:
Safety and Tolerability Assessment of Adcetris
Assessmentof Immunogenicity
MMAE metabolite identification
4) Clinical DDI study design
4.1) Grouping: three dosing groups, open-label, phase I trial, 2 dosing (2 dosing cycles) 21 days apart;
a) Midazolam co-administration group: Adcetris was administered IV at 1.8 mg/kg on the first day of dosing cycles 1 and 2, respectively, and midazolam was administered on the first and last three days of dosing cycle 1, respectively Administer 1 mg, and collect blood until 24 hours on the first three days and the last three days of dosing cycle 1;
b) Rifampicin co-administration group: Adcetris was administered IV at 1.8 mg/kg on the first day of dosing cycle 1 and 2, respectively, from day 14 of dosing cycle 1 to day 21 of dosing cycle 2 Up to now, 600 mg of rifampicin was administered orally every day, and blood was collected several times on the first day of each dosing cycle, once a day on the 2nd to 5th day, and once every 2 weeks after 3 weeks;
c) Concomitant ketoconazole group: Adcetris was administered IV at 1.2 mg/kg on the first day of dosing cycle 1 and 2, respectively, from day 19 of dosing cycle 1 to day 21 of dosing cycle 2 400 mg of ketoconazole was administered orally every day, and blood was collected several times on the first day of each administration cycle, once a day on the 2nd to 5th day, and once every 2 weeks after 3 weeks;
d) The rifampicin co-administration group was used to evaluate the excretion route of MMAE: feces and urine were collected for one week from the start of Adcetris administration to the time period before administration of rifampicin;
4.2) Number of cases: 12 cases in each group, a total of 36 patients;
4.3) Assessment parameters:
Pharmacokinetic parameters: plasma concentrations of Adcetris, CYP3A4 drugs, free MMAE and total antibody concentrations
safety
4.4) Data analysis: Cmax, Tmax, AUC, Vss, CL, elimination half-life…
5) Results of clinical DDI studies
Adcetris had no effect on the PK of midazolam;
Rifampicin had no effect on ADC exposure itself, but reduced MMAE exposure by 46%;
Ketoconazole had no effect on ADC exposure per se, but increased MMAE exposure by 34%.
6) The description of the DDI part in the contents of the manual
6.1) Intensive detection of adverse reactions is required when combined with strong CYP3A4 inhibitors;
6.2) In vitro studies show that MMAE is a substrate and inhibitor of CYP3A;
6.3) Effects of marketed drugs on Adcetris:
MMAE is mainly metabolized by CYP3A. When combined with ketoconazole, a strong CYP3A4 inhibitor, the exposure of MMAE will increase by 34%. Therefore, patients need to intensively detect adverse reactions when taking CYP3A4 inhibitors at the same time; CYP3A4 strong inducer rifampicin will cause MMAE exposure decreased by 46%.
6.4) Effects of Adcetris on marketed drugs:
Adcetris did not affect exposure to the CYP3A4 substrate drug midazolam. MMAE also has no inhibitory effect on other CYP enzymes at clinical plasma concentrations. Therefore, Adcetris is considered to have no effect on exposure to CYP3A4 substrate drugs.
Summarize
1) The antibody part is designed according to the macromolecular DDI guidelines and the mechanism of action of the antibody itself;
2) Toxin small molecules follow guidelines based on metabolic enzymes and transporter DDI:
Listed small molecules can directly refer to the application materials;
A full set of in vitro DDI assays are theoretically performed on new small molecules, focusing on the research of metabolic enzyme transporter substrates;
The DDI risk of small molecule active metabolites of toxins needs to be investigated;
If it is a substrate, clinical DDI studies need to be considered, which can be combined with other clinical pharmacology trials.
References
1.FDA.Drug-Drug Interaction Assessment for Therapeutic Proteins Guidance for Industry.
2.FDA.Clinical Pharmacology Considerations for Antibody-Drug Conjugates.
3. Kraynov E et al. Current Approaches for ADME Characterization of Antibody-Drug Conjugates: An Industry White Paper. Drug Metabolism and Disposition. 2015, 15.
4. Mahmood I. Effect of Intrinsic and Extrinsic Factors on the Pharmacokinetics of Antibody-Drug Conjugates (ADCs). Antibodies. 2021, 10 (40).
5. Lievano FA et al. Risk Minimization of Antibody-Drug Conjugates in Oncology: A Review. Drug Safety. 2021.
6. ADCETRISTM (brentuximab vedotin) for Injection For intravenous infusion. FDA Label.
7. ADCETRISTM (brentuximab vedotin) non-clinical and clinical application materials. PMDA.