On August 24, BioMarin announced that the European Union approved the conditional marketing of Roctavian (Valoctocogene roxaparvovec) gene therapy for the treatment of adults with severe hemophilia A who do not have coagulation factor VIII inhibitors and adeno-associated virus 5 (AAV5) antibodies. patient. Roctavian is the first approved gene therapy for hemophilia A. In addition, the European Union maintained orphan drug designation for the therapy and granted it a 10-year market exclusivity period.
Hemophilia A is a hereditary disease caused by coagulation dysfunction due to coagulation factor VIII (FVIII) deficiency. It is also the most common type of hemophilia. The incidence is higher in men. The clinical manifestations are bleeding from any part of the body, including joints, Muscle and deep tissue. The degree of bleeding in patients with hemophilia A is mainly determined by the level of FVIII activity, and patients with severe hemophilia A (residual FVIII activity <1%) account for 50% of the total patient population. At present, the standard treatment for hemophilia A is 2-3 times a week of preventive intravenous infusion of FVIII, and the total number of injections per year is as high as about 104-183 times. The burden of treatment is self-evident. Even with this prophylactic regimen, patients are still at risk of spontaneous bleeding.
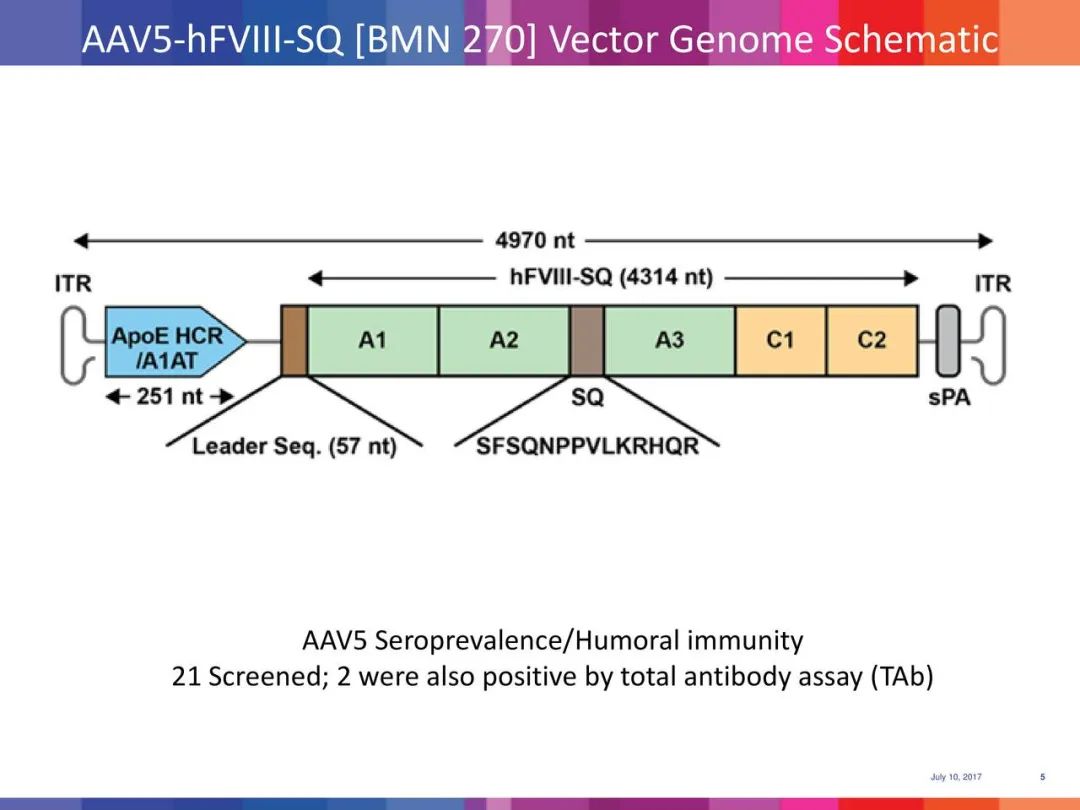
Valoctocogene roxaparvovec is a gene therapy that uses an AAV5 viral vector to deliver the FVIII transgene. The advantage of this therapy is that it may only require a single treatment to obtain the gene expressing FVIII, thus eliminating the need for long-term prophylactic coagulation factor injections.
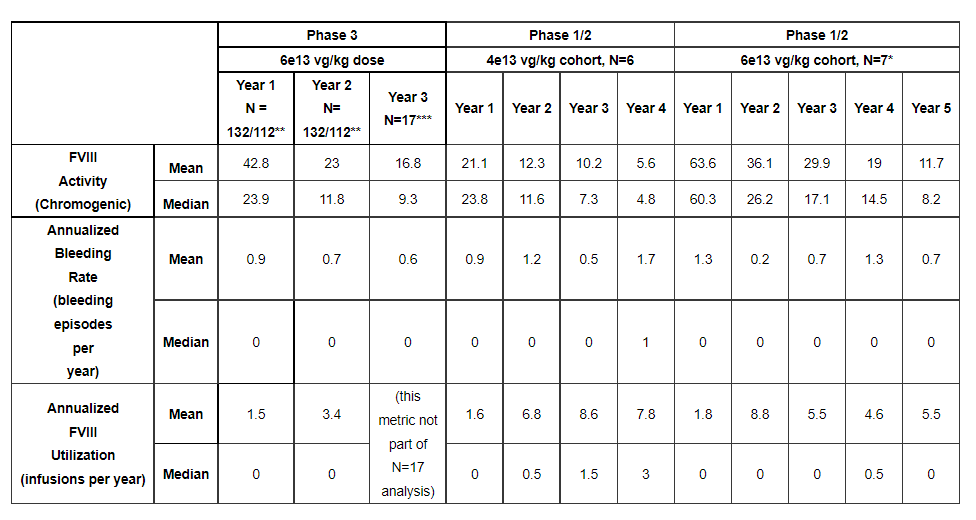
The approval is based on substantial evidence, including 2-year follow-up data from the GENEr8-1 study. GENEr8-1 is a single-arm, multicenter, open-label study and the largest phase III gene therapy study in hemophilia to date, evaluating Valoctocogene roxaparvovec in patients with severe type A previously treated with FVIII prophylaxis. Efficacy and safety in patients with hemophilia. A total of 134 patients were enrolled in the study. The primary endpoint was the change from baseline in median FVIII activity levels between 49 and 52 weeks after treatment, and the secondary endpoints were the average annual use of exogenous FVIII replacement therapy and annualized bleeding. Rate.
RESULTS: Among 132 HIV-negative patients, mean FVIII activity increased by 41.9 IU/dL (95% CI: 34.1-49.7; median change: 22.9 IU/dL) at weeks 49-52. Among the 112 patients enrolled in the prospective non-intervention study, the mean annualized use rate and annualized bleeding rate of FVIII after week 4 of infusion decreased by 98.6% and 83.8%, respectively (P<0.001). Among them, the mean annualized bleeding rate was 0.9 (median: 0.0) in the first year and 0.7 (median: 0.0) in the second year, demonstrating the stable and durable bleeding control effect of the therapy.
The safety profile of Valoctocogene roxaparvovec was favorable, with no patients experiencing FVIII inhibition or thromboembolic events. During the second year of follow-up, no new safety signals emerged. Elevated ALT levels occurred early in treatment in 119 patients (89%), most of which were controlled with corticosteroids. Other common adverse events included headache (41%), nausea (38%), and increased AST (35%).
Although Valoctocogene roxaparvovec is the world’s first gene therapy for hemophilia to enter the market review stage, its road to market is not smooth. BioMarin submitted marketing applications for the therapy to the EMA and the FDA in 2019, but was rejected by the FDA in August 2020, and was advised to use the 2-year follow-up annualized bleeding rate as the primary endpoint to evaluate the durable benefit of the therapy . In November of the same year, BioMarin withdrew its European marketing application for the therapy.
In January 2021, BioMarin announced positive data from the 1-year follow-up of the GENEr8-1 study, and submitted a marketing application to the EU again in June. In January of this year, positive data from the GENEr8-1 study with a 2-year follow-up were disclosed. BioMarin plans to resubmit its marketing application to the FDA by the end of September.