The design of clinically successful ADCs depends not only on the potency of the payload and its point of attachment, linker stability, and efficient drug release, but also on the choice of antibody and bioconjugation technology. All FDA-approved ADCs over the past 10 years have been in the form of heterogeneous mixtures of ADCs with varying amounts of drug attached to different positions of the mAb. The conjugation site has a significant effect on ADC stability and its pharmacokinetics, with high DAR (drug-to-antibody ratio) often resulting in rapid plasma clearance, while low DAR ADCs exhibit weaker activity. Among ADC drugs, the presence of naked mAb is a potent competitive inhibitor. Thus, over the past decade, a host of new conjugation strategies have been developed with the aim of controlling the location and quantity of small-molecule drugs while maintaining structural integrity and homogeneity.
Chemical-based specific in situ antibody modification
The natural structure of monoclonal antibodies offers multiple possibilities for biological conjugation, and chemical-based, specific conjugation of natural (non-engineered) antibodies has several advantages. It avoids the complexities of site-specific mutation of antibodies and possible challenges in scale-up and optimization of cell cultures.
Conjugation Sites Depending on the antibody sequence, the inter-disulfide attachment sites for endogenous amino acids such as lysine, histidine, tyrosine, and cysteine are very attractive. All FDA-approved ADCs until 2021 utilize these endogenous amino acids for conjugation. However, antibody scaffolds also contain glycans, which result from post-translational modifications of the FC region during monoclonal antibody production. Several studies have reported new strategies for glycoengineering, which appears to be an interesting alternative to bioconjugation.
Conjugation to endogenous amino acids
One of the most common conjugation methods utilizes the lysine residue of an antibody, where the amino acid nucleophilic NH2 group reacts with an electrophilic N-hydroxysuccinimide (NHS) group on the Lick payload. Despite the simplicity of the reaction, the high abundance of available lysine residues leads to the formation of a heterogeneous mixture of many ADCs under random distribution. DAR is controlled by drug/antibody stoichiometry, and this approach is widely used, including approved ADCs such as Besponsa, Mylotarg, and Kadcyla.
More recently, specific modifications to lysine sites and residues have also been reported. By computer-aided design, sulfonyl acrylates were used as intermediate reagents for modification of single lysine residues on native protein sequences.
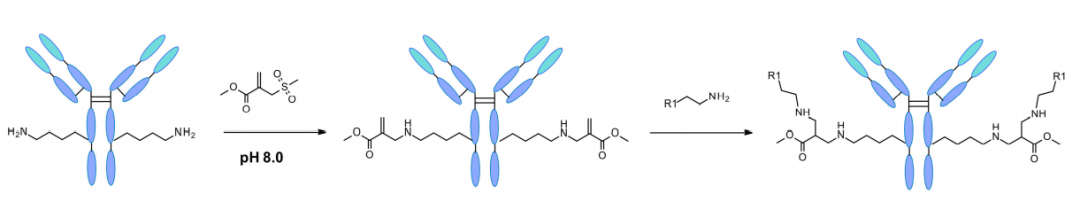
The regioselectivity of the reaction is attributed to the design of the sulfonyl acrylate and the unique local microenvironment surrounding each lysine. It was computationally predicted that lysines with the lowest pKa readily react preferentially at weakly basic pH in a site-specific manner. This response was observed even in the presence of other nucleophilic residues such as cysteine. The technology has been applied to 5 different proteins and trastuzumab, all retaining the original secondary structure and protein function after conjugation.
In 2018, Rai et al. reported another site-specific modification utilizing reversible intermolecular reactions of “chemically key proteins”. This reagent carries a variety of functional groups that reversibly form imine moieties on all available lysine residues. The key protein is then reacted with the proximal histidine residue via the epoxide in the reagent. Thus, under physiological conditions, key proteins are dissociated from lysines and aldehydes are regenerated to enable labeling of antibodies by oxime binding.
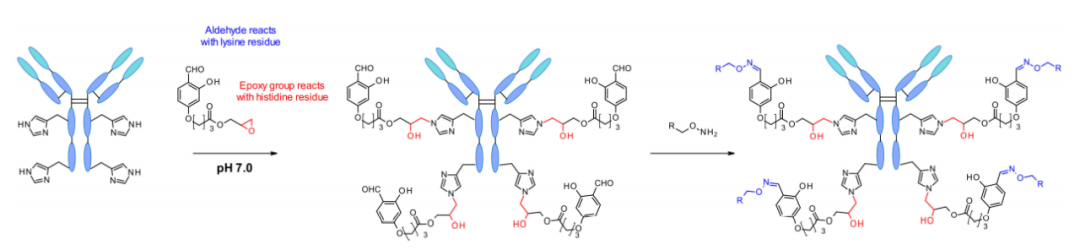
This technology for targeted modification of key proteins was later developed into single-lysine residue labeling with unquestionable selectivity even in the presence of N-terminal amines. The success of the method relies on the Fk1-spacer-Fk2 reagent.
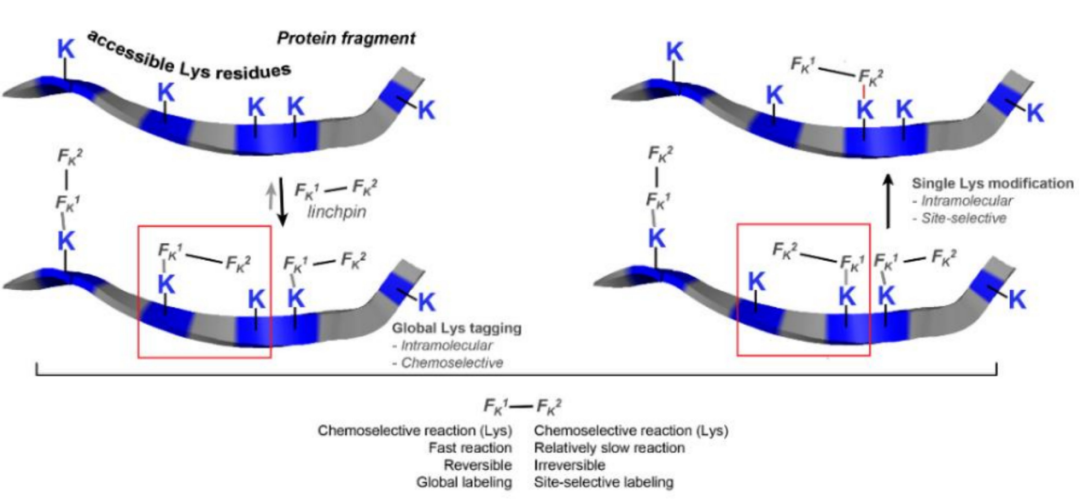
The Fk1 functional group reacts reversibly with lysine to modulate the microenvironment of the proximal lysine moiety of Fk2. Coupling was then carried out via an amide bond at the lysine residues (K169 and K395) at Fk2, and the design of the spacer modulates the position of the coupling. This method has been successfully applied to the synthesis of ADC (trastuzumab-emtansine), demonstrating that its cellular activity is comparable to the approved Kadcyla.
Merlul et al. recently reported a different conjugation strategy that efficiently targets histidine residues on native antibodies. They introduced a cationic organometallic platinum(II)-based linker, [ethylenediamineplatinum(II)]2+, denoted Lx in the figure.
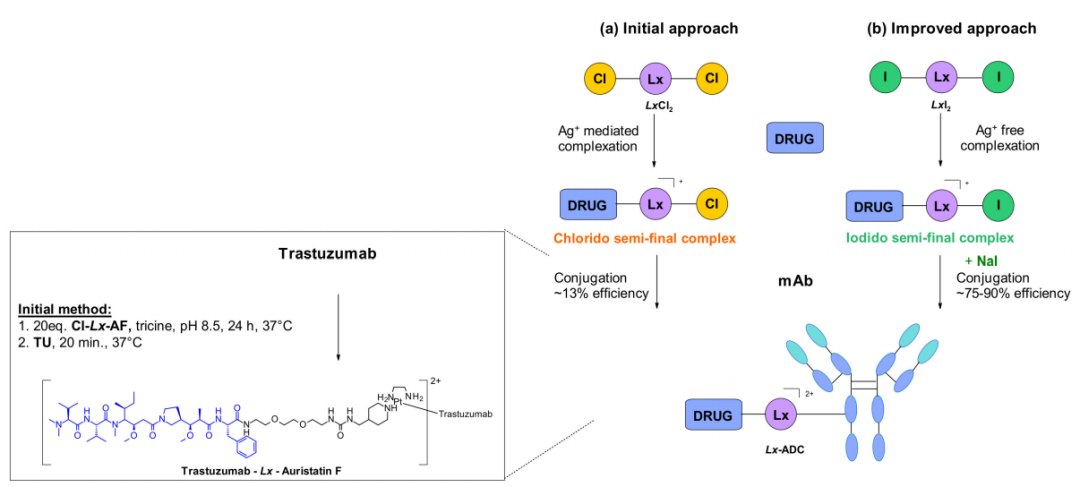
This technique is based on two steps of complexation and coupling. A nitrogen-heterocyclic ligands such as piperidine coordinate with Lx to form complex precursors, and stable intermediates contain the payload and a chloride ion on the ligand. This complex contains a positively charged Pt(II) center, which improves the water solubility of the linker and payload complexes and minimizes antibody aggregation, and this approach is also extended to similar iodine complexes. In a recent report, the use of sodium iodide was shown to significantly improve the coupling yield and selectivity of this technique. The exchange of the residual chloroligand on the Cl-Lx-drug-loaded complex with the iodide generates a more active I-Lx-drug-load, resulting in higher coupling yields. This technology has been applied to the large-scale production of ADC drugs.
Disulfide re-bridging strategy
IgG antibodies contain four interchain disulfide bonds, two connecting the light and heavy chains, and two in the hinge region connecting the two heavy chains, which maintain the integrity of the monoclonal antibody. Another classical bioconjugation pathway explored the role of these cysteines as payload attachment points. Reduction of the four disulfide bonds typically yields eight sulfhydryl groups that are able to react with the maleimide linker, resulting in an ADC with a DAR of 8.
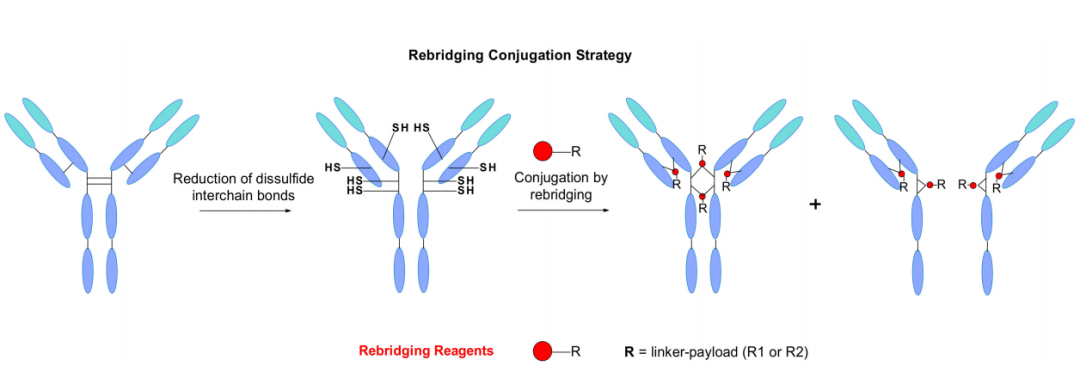
Doronina and colleagues report an example of an ADC with a chimeric anti-CD30 monoclonal antibody conjugated to MMAE with a DAR=8. This way of payload loading is better controlled than classical lysine coupling. However, higher drug loads have been reported to increase the risk of aggregation, resulting in high plasma clearance and reduced efficacy in vivo.
A new site-specific re-bridging conjugation strategy was reported by Badescu and al in 2014, and they were the first to demonstrate that new bis-sulfones can alkylate reduced disulfides from antibodies and antibody fragments The two sulfhydryl groups of the bond have the least effect on antigen binding. Later, Wang and al described a new water-soluble allyl sulfone that enhanced reactivity without in situ activation. It exhibits high stability, high water solubility and site specificity.
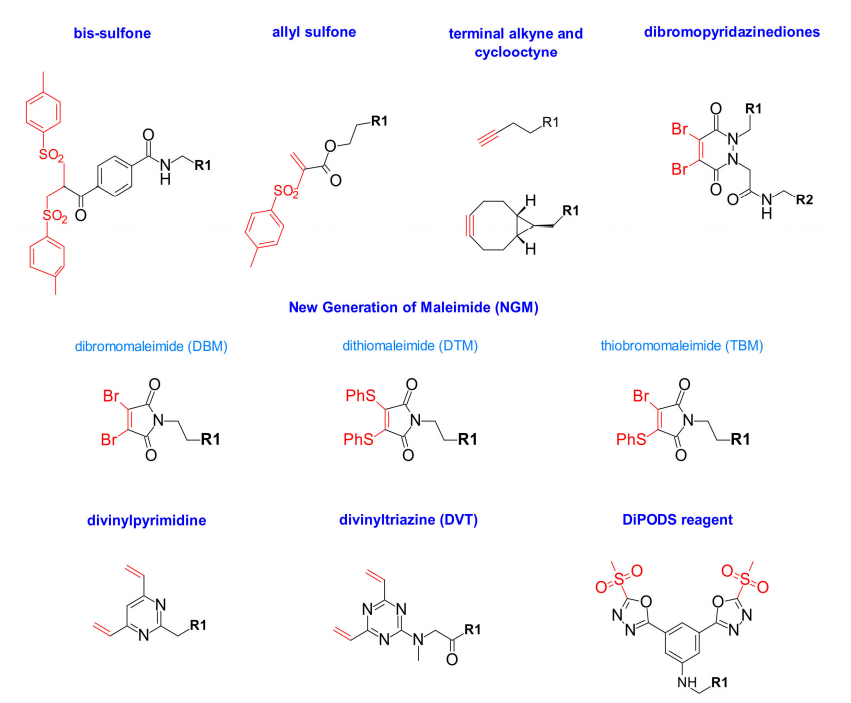
In addition, there are re-bridging technologies for the bioconjugation of mercaptoalkynes with terminal alkynes and cyclooctynes, which further develops a new generation of maleimides such as dibromo-(DBM) and dithiomaleimides (DTM) for site-specific conjugation. These maleimide analogs contain good leaving groups at positions 3 and 4, enabling fast, efficient, and high-yield coupling. Hybrid thiobromomaleimides (TBMs) that combine dibromo and dithiomaleimide properties have recently been reported. This TBM reagent binds faster and shows a higher percentage of DAR=4, This may be due to the reduction of steric hindrance by bromine.
In 2015, Chudasama et al. introduced a new class of heavy bridging reagents, dibromopyridazinediones. They demonstrated efficient insertion into disulfide bonds, and the resulting structure exhibited excellent hydrolytic stability even at high temperatures. However, heterogeneity was also observed with increasing temperature on the reduction step, and this structure also allows for the selective introduction of different functional groups.
Divinylpyrimidine is another potent re-bridging reagent that produces stable ADCs with DAR=4. The effect of vinylheteroaryl scaffolds on cysteine re-bridging was investigated by Spring et al., who suggested that replacing pyridine with pyrimidine could make the heteroaryl ring a better electron acceptor, thereby increasing the crosslinking efficiency. Extending their work to divinyltriazines, heavy bridging showed higher efficiency at high temperatures.
To avoid the drawbacks of in vivo instability associated with classical maleimide coupling, Barbas et al. investigated methylsulfonylphenyloxadiazole, a reagent that reacts specifically with cysteine. They are more stable than cysteine-maleimide conjugates in plasma. Inspired by this, Zeglis designed the DiPODS reagent, which contains two oxadiazolyl methyl sulfone moieties linked by phenyl groups, and DiPODS forms a covalent bond with two sulfate radicals in a re-bridging manner. Coupling in this way has superior in vitro stability and in vivo performance compared to maleimide coupling.
Glycan conjugation
Since IgG is a glycoprotein, it contains an N-glycan at position N297 of the CH2 domain of each heavy chain of the Fc fragment, and this glycosylation can serve as an attachment point for the payload. The long-distance localization of the polysaccharide to the Fab region reduces the risk of compromising the antigen-binding capacity of the antibody after conjugation, and in addition, their chemical composition is different compared to the peptide chain of the antibody, allowing site-specific modifications, making them suitable the coupling site.
Glycan bioconjugation can be differentiated according to the techniques used to target carbohydrates: including glycan metabolic engineering, glycotransferase treatment after glycan oxidation, ketone or azide labeling after endoglycosidase and transferase treatment.
Neri et al. reported site-specific modification of fucose at the N-glycosylation site of IgG antibodies. This sugar contains a cis-diol moiety suitable for selective oxidation. They oxidized the fucose residue with sodium metaperiodate to create an aldehyde group capable of reacting with a hydrazine-containing linker, so that the antibody was linked to the drug through a hydrazone bond.
Senter and colleagues added sulfur-based analogs to cell culture media to metabolize 6-thiofucose into antibody modifications. They believe that the substitution is accomplished by hijacking the fucosylation pathway, thus introducing chemical sites for site-specific binding. Compared to classical cysteine conjugates, this approach significantly reduces the level of heterogeneity and yields conjugates with more predictable pharmacokinetic and pharmacodynamic properties.
Sialic acid is rarely present in recombinant IgG, however, it has been demonstrated that glycine can be enzymatically engineered using galactosyl and sialyltransferases. Galactose was added by an enzymatic reaction to obtain G2 glycans, followed by the addition of terminal sialic acids. This modification generates an aldehyde group by periodic acid oxidation, which can be coupled to a linker-payload with a hydroxylamine group. The obtained conjugate has high targeting selectivity and good antitumor activity in vivo. Periodic acid can also oxidize sensitive amino acids such as methionine and affect the binding to FcRn.
In addition to these conjugation strategies, galactose residues can also serve as modification sites. Several studies have reported the use of mutant β-1,4-galactosyltransferases to replace galactose with a galactose containing a ketone or azide functional group, a galactose derivative with biorthogonal functional groups as Efficient conjugation opens the way. These techniques have been developed for imaging and anticancer applications.
The endoglycosidases EndoS and EndoS2 were discovered from Streptococcus pyogenes. These enzymes are able to hydrolyze the N-glycans of IgG, making the hydrolyzed residues effective sites for bioconjugation. This method helps to homogenize the glycan structure of the mAb, and it also works for any IgG subtype. Such methods were applied to trastuzumab-maytansine to prepare glycoconjugated ADCs with good in vitro and in vivo efficacy.
Site-specific bioconjugation of engineered antibodies
Advances in the fields of bioorthogonal chemistry and protein engineering are helping to generate more homogeneous ADCs. Although there are many available attachment methods on native mAbs, site-specific bioconjugation on engineered antibodies enables more efficient DAR control and avoids changing the affinity for antigen binding. In this way, natural or unnatural amino acids are added at certain positions, resulting in a homogeneous product with excellent pharmacokinetic and pharmacodynamic characteristics.
Enzymatic method
Attachment of the payload can be achieved in a very selective manner by inserting specific amino acid tags into the antibody sequence. These tags are recognized by specific enzymes, such as formylglycine-generating enzymes (FGE), microbial transglutaminase (MTG), transpeptidases, or tyrosinase, enabling site-specific conjugation.
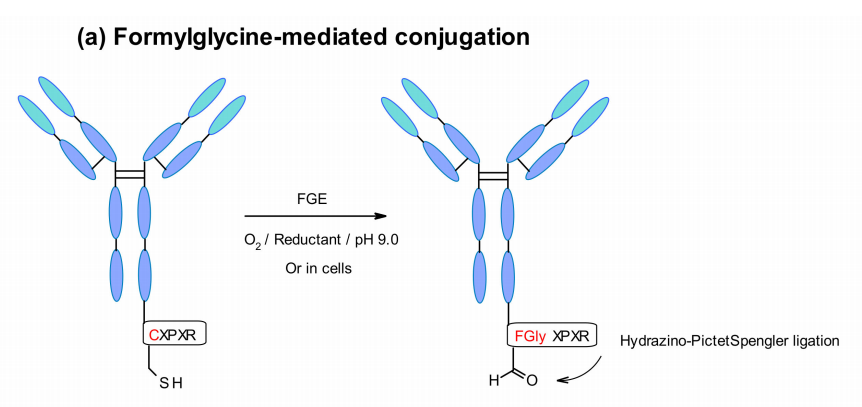
Aaron et al. explored a new site-specific conjugation using aldehyde-tagged proteins. The technique utilizes a genetically encoded pentapeptide sequence (Cys-X-Pro-X-Arg) in which cysteine residues are recognized by FGE and co-translationally oxidized to formylglycine during protein expression in cells. In this way, engineered antibodies are selectively coupled to aldehyde-specific linkers via HIPS (hydrazino-Pictet-Spengler) chemistry.
Microbial transglutaminase (MTGase) strategies are also frequently developed for site-specific conjugation. MTGase catalyzes the formation of a peptide bond between the glutamine side chain at position 295 of the deglycosylated antibody and the primary amine of the substrate. In contrast to other enzymatic strategies, MTG is a flexible technique that does not require a peptide donor for conjugation. There are no structural limitations as long as the acyl acceptor contains a primary amine.
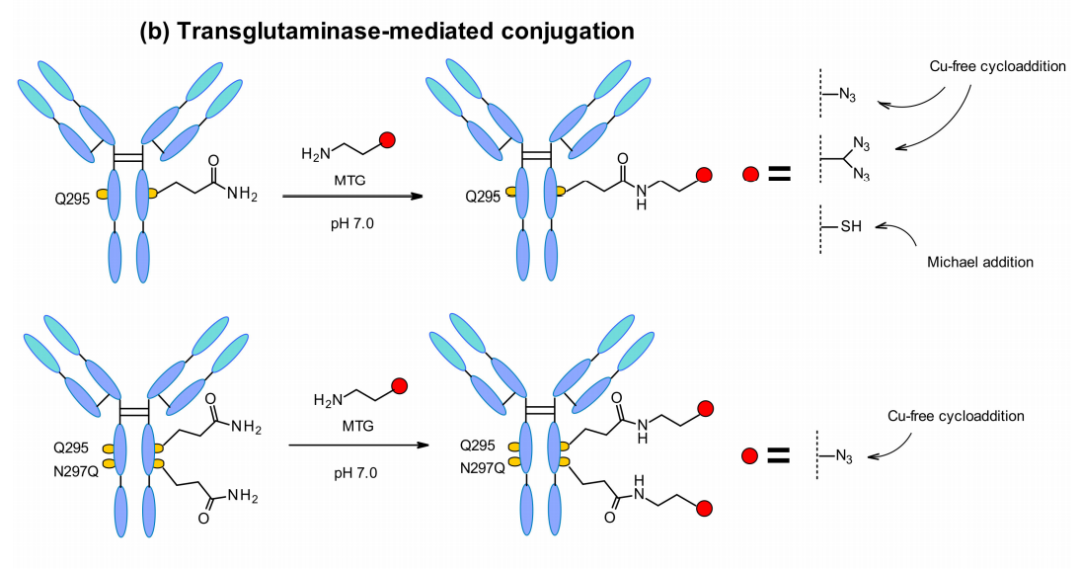
Glutamine residues are naturally present in the Fc region of each heavy chain of mAbs. After deglycosylation at position 295, the glutamine residues are coupled through an MTGase-mediated reaction, resulting in a homogeneous ADC with DAR=2. To improve efficiency, a branched linker can be coupled to double the DAR, and mutation of the asparagine to glutamine at position 297 also increases the DAR.
NBE Therapeutics developed S. aureus transpeptidase A-mediated conjugation. Their strategy utilizes transpeptidase A (SrtA), which cleaves the amide bond between threonine and glycine residues in the motif of the LPXTG (X = any amino acid) pentapeptide. It then catalyzes the coupling of the glycine-related payload to the newly generated C-terminus, generating peptide bonds at physiological temperature and pH.
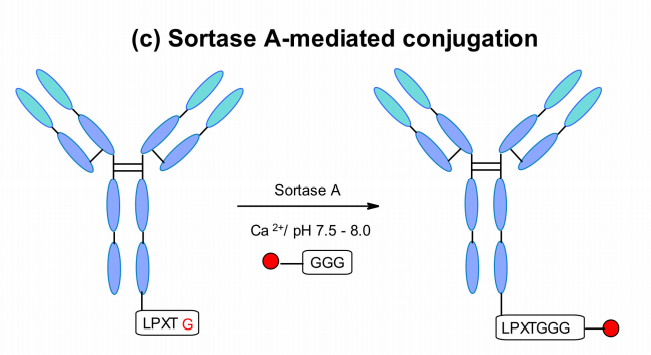
This approach was applied to different antibodies, such as anti-CD30 and anti-Her2, and conjugated with maytansine and MMAE using a 5-glycine-labeled linker, and both ADCs showed similar in vitro cell-killing activity to classical conjugation. The enzymatically produced trastuzumab-maytansine matched Kadcyla exactly in vivo.
In another example, an ADC of the highly potent anthracycline toxin derivative PNU-159682 was generated using a transpeptidase method. Interestingly, with this technique, the coupling efficiency was even higher than the Adcetris and Kadcyla analogs. Furthermore, the as-prepared PNU-159682 ADC exhibited high in vitro and in vivo stability and demonstrated potency over ADCs containing tubulin-targeting payloads.
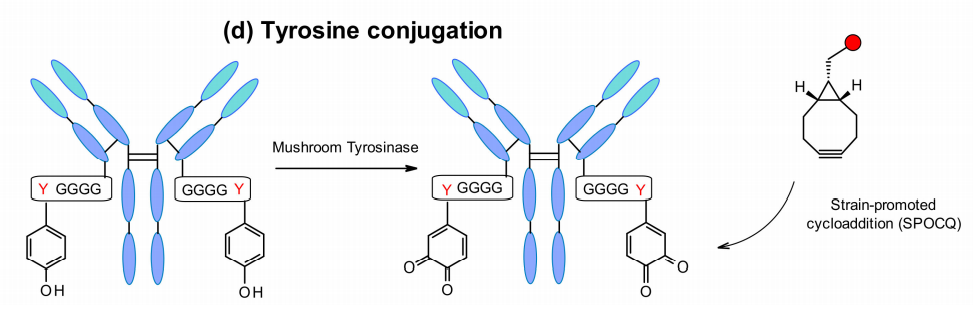
Another emerging new approach is site-specific antibody labeling via a tyrosine tag that is genetically fused to the C-terminus of the monoclonal antibody light chain. Considering site accessibility, Bruins and colleagues used an engineered tetraglycyl tyrosine residue as a tag, which provides an easily accessible site for conjugation. Tyrosinase oxidizes tyrosine to 1,2-quinone, allowing cycloaddition reactions with various bicyclo[6.1.0]nonyne (BCN) derivatives. This method allows efficient coupling with MMAE containing a BCN linker.
Cysteine Engineering: Thiemab Technology
Random cysteine conjugation and re-bridging are techniques that utilize naturally occurring cysteine residues within the antibody structure. However, the heterogeneity of the random cysteine approach and the fragmentation of mAbs in re-bridging strategies need to be considered in ADC synthesis, especially when hydrophobic drugs are conjugated.
Unlike them, thiimab technology enables selective and uniform modification of desired sites on antibodies by utilizing engineered reactive cysteines that do not involve structural disulfide bonds. Generally, cysteine mutations are designed to facilitate cytotoxic payload conjugation while maintaining mAb stability, affinity, and minimizing ADC aggregation. To determine the optimal location of mutations, several techniques are commonly employed, including computational modeling, model system screening, and high-throughput scanning.
Junutula et al. first reported a thiomab strategy in which alanine at position 114 of the anti-MUC16 antibody heavy chain (HC-A114) was replaced with an engineered cysteine residue, a reactive thiol within the engineered position Capable of reacting with maleimide-loaded linkers. Synthetic anti-MUC16 ADCs demonstrated potency in xenograft mouse models and high dose tolerance in rats and cynomolgus monkeys, a finding that establishes a general approach to thiimumab conjugation strategies.
In addition, succinimide linkage can undergo two parallel reactions in the cytoplasm: a reverse Michael reaction leading to loss of linker-payload, and hydrolysis of succinimide, both of which have significant effects on ADC activity in vivo influences. To improve stability, Lyon and collaborators designed a linker that integrates with the basic amino group adjacent to the maleimide. The addition of diaminopropionic acid (DPR) to the linker promotes rapid quantitative hydrolysis of sulfosuccinimide at neutral pH and room temperature, in this way, non-specific decoupling is prevented, resulting in improved in vivo stability . Besides the commonly used maleimides, different cysteine reactants such as iodoacetamide, bromoformamide, carbonyl acrylates, N-alkyl vinyl pyridine salts were also explored.
Bioconjugation with engineered unnatural amino acids
In addition to thiimab technology, the addition of non-standard amino acids (ncAA) provides another possibility for site-specific conjugation. The technology uses amino acids with unique chemical structures that enable the introduction of linker-payload complexes in a chemoselective manner. This technique requires recombination of antibody sequences, utilizing tRNAs that are orthogonal to all endogenous tRNAs and synthetases in the host cell, and aminoacyl tRNA synthetases (aaRS) for bringing ncAAs into proteins in response to unassigned codons. Typically, ncAA is added to the medium during fermentation. The choice of unnatural amino acids is important because they may provoke immunogenicity. Commonly used ncAAs are analogs of natural amino acids with unique groups such as ketones, azides, cyclopropenes or dienes.
Previous studies have successfully incorporated p-acetylphenylalanine (pAcF) into anti-CXCR4 antibodies. The payload, Auristin, is efficiently conjugated to the antibody via an oxime linkage, resulting in a chemically homogeneous ADC. The ADC showed good in vitro activity and complete clearance of lung tumors in mice.
Due to the acidic conditions required for oxime attachment and the kinetics of slow ADC release, another option is to add ncAA-containing azides. The widely available p-azidofenilide (pAzF) can rapidly carry out CuAAC or SPAAC reactions under physiological conditions, and this strategy was used to successfully couple glucocorticoid payloads on anti-CD74 antibodies. In addition to pAcF technology, azide-containing lysine analogs (AzK) were successfully incorporated into antibodies to generate site-specific ADCs with Auristin, PBD dimer, or tubulin payloads.
Furthermore, cyclopropene derivatives of lysine (CypK) as well as naturally occurring atypical amino acids such as selenocysteine (Sec) were successfully incorporated into antibodies. The resulting ADCs exhibited good stability, selectivity, and in vitro and in vivo activities.
Over the past few years, many advances have been made in structural optimization and mechanistic expansion of ADCs. New conjugation techniques have been developed to obtain higher selectivity for tumors. These conjugation techniques enable ADCs with better stability, selectivity, and in vitro and in vivo activity. The preliminary data of these novel conjugation technologies are encouraging and will greatly facilitate the rapid development of ADC drugs in the future.