Antibody-drug conjugate (ADC) therapy has developed rapidly in recent decades. Currently, 14 products have been approved worldwide and more than 140 ADCs are in clinical trials. By 2030, the ADC market will reach over $15 billion. Rationale for ADCs: By combining the specificity of monoclonal antibodies with the cytotoxicity of potent small-molecule drugs, ADCs can precisely deliver toxins to tumors.
However, despite the FDA approval of multiple ADC drugs, the failure rate of ADCs remains high during clinical development. The inherent complexity of ADCs is a double-edged sword that provides opportunities for better treatment while also increasing confounding factors for treatment failure. ADC design drives its pharmacokinetics and pharmacodynamics, and requires more in-depth analysis than the commonly used Cmax and area under the curve (AUC) metrics to obtain optimal doses for clinical application. Current FDA-approved ADCs targeting solid tumors share some common features, including humanized IgG1 antibody domains, highly expressed tumor receptors, and high-dose antibodies. These common features have potential implications for clinical pharmacokinetics and mechanism of action, and provide reference for ADC design at all stages of clinical development.
3 Design Criteria Followed by Recently Approved ADCs
Three recently approved solid tumor ADCs underscore important design criteria. Although several components of the ADC show significant differences, their shared properties are worth noting. The structures of the four FDA-approved ADCs for solid tumors are very different, including different linker types (cleavable vs. non-cleavable, different release mechanisms, different stability), specific and non-specific binding, different targets , cancer type and drug-to-antibody ratio (DAR). Interestingly, these ADCs share three characteristics: (i) high target expression (105–106 receptors/cell), (ii) high antibody dose (3.6 mg/kg or greater over 3 weeks), and (iii) IgG1 isotype antibody backbone
Table 1.
Structural components for the FDA approved ADCs for solid tumor indications. These ADCs display differences in selected payload, linker, DAR, and conjugation. In contrast, the antibody isotype, large doses, and high expression target are shared for these therapies.
| FDA approved ADCs for solid tumors (year) | Kadcyla (2013) | Padcev (2019) | Enhertu (2019) | Trodelvy (2020) |
|---|---|---|---|---|
|
|
||||
| Target | Her2 | Nectin-4 | Her2 | Trop-2 |
| Antibody isotype | IgG1 | IgG1 | IgG1 | IgG1 |
| Clinical dose over 21 days* (mg/kg) |
3.6 | 3.75 | 5.4 | 20 |
| Linker | Non-cleavable (SMCC) | Cleavable (VC) | Cleavable (tetrapeptide) | Cleavable (CL2A) |
| Payload | Microtubule inhibitor (DM1) | Microtubule inhibitor (MMAE) | Topoisomerase inhibitor (Exatecan derivative) | Topoisomerase inhibitor (SN-38) |
| Drug to antibody ratio | 3.5 | 4 | 8 | 7.6 |
| ADC clearance half-life (days) | 4 | 3.4 | 5.7 | 0.67 |
| Payload clearance half-life (days) | Not measured | 2.4 | 5.8 | 0.75 |
These three common characteristics have major implications for drug delivery and distribution. In fact, since ADCs use known cytotoxic payloads (such as microtubule inhibitors) and known targeting antibodies, a key feature of their clinical success is delivery—targeting each at tolerable doses The payload of tumor cells. These shared design features have individual implications for tumor-targeted delivery of payloads.
Highly expressed target
HER2, Nectin-4 and Trop-2 are highly expressed tumor antigens, with more than 105 receptors per tumor cell, and their expression is significantly reduced in healthy tissues. Due to the large expression differences, highly expressed targets can provide a larger therapeutic window. Because drug delivery to healthy tissues is often more efficient than delivery to tumors, high antibody doses and high expression of tumor targets can rapidly saturate uptake in low-expressing healthy tissues, while still maximizing tumor uptake. Payload toxicity and/or DAR can then be modified to ensure delivery to tumor cells above the therapeutic threshold, while maintaining sub-therapeutic thresholds in healthy tissue (to avoid target-mediated healthy tissue toxicity). In contrast, targeting less expressed tumor antigens requires a more potent payload to reach therapeutic concentrations in targeted cells. Increasing payload potency generally results in higher toxicity and lower ADC tolerated doses. While lower ADC doses reduce tumor uptake, but may not reduce healthy tissue uptake by the same amount (eg, if target-mediated healthy tissue uptake remains saturated), potentially lowering the therapeutic index. It is worth noting that this trade-off is very different from small-molecule drugs, which are generally balanced with plasma concentrations, so lower doses result in lower exposure to healthy tissue. In stark contrast to small molecules, lower doses of more potent ADCs can limit tumor penetration, thereby reducing efficacy rather than toxicity.
high antibody dose
The development of ADCs targeting solid tumors is challenging because of their leaky blood vessels, tortuosity, and poor lymphatic drainage, leading to poor convection and elevated interstitial pressure. These features combine to create an unfavorable environment for the delivery of ADC drugs. The most direct way to increase antibody delivery and tissue penetration is to administer higher doses of the antibody, the second shared feature among currently approved ADCs.
Kadcyla is the first ADC to receive FDA approval for solid tumors in many years, and was approved in 2013 by the use of a human IgG1 backbone, a moderate DAR (3.5), a non-cleavable linker, and a potent microtubule inhibitor. Many current ADCs targeting solid tumors have higher doses (3.75-20 mg/kg) than Kadcyla at 3 weeks, significantly exceeding many approved ADCs targeting hematological tumors (eg, Besponsa and Zynlonta at doses of ~0.02, respectively) and 0.15 mg/kg). These doses are necessary to overcome high expression and efficient internalization to deliver the payload to cells.
Other design criteria
The choice of payload is critical to ADC development. Today, most payloads fall into one of three categories: (i) DNA damage inducers, (ii) microtubule inhibitors, and (iii) topoisomerase inhibitors. DNA damage payloads are often very potent (eg PBD), while microtubule (DM4, MMAE) and topoisomerase (exatecan, SN-38) inhibitors are milder. Determining the optimal payload requires analysis on a case-by-case basis. Lower payloads provide greater maximum tolerated dose (MTD). However, for indications with lower antigen presentation, the delivery of the payload may not exceed the therapeutic threshold, thus requiring a higher potency payload. In addition to potency, recent studies have identified multiple payloads as inducers of immunogenic cell death (ICD). The ability of some payloads to elicit an immune response following cell death is a new avenue of research that may have broad implications for next-generation ADC payload selection.
The choice of linker is also critical, non-cleavable linkers are generally more stable in plasma. Significant progress has also been made in linker chemistry. For example, the Kadcyla non-cleavable linker lost 18.4% of its payload in 4 days, while the Enhertu cleavable linker lost 2.1% of its payload in 21 days. However, even if linkers can reduce ADC payload loss in the circulation, more difficult challenges are encountered following systemic uptake and degradation of ADCs themselves. Since most ADC doses do not reach the tumor, the ADC will be metabolized elsewhere in the body, releasing the payload at undesired locations. Payloads, linkers, and binding sites can all influence where in vivo nonspecific release occurs and dose-limiting toxicity (DLT).
Current FDA-approved drugs exhibit significant variability in these design features, suggesting the need to personalize ADCs for their specific targets. However, the similarity of high-dose antibodies and highly expressed targets, combined with preclinical evidence, suggest that tissue penetration and tumor saturation are key components of efficacy in solid tumors. Therefore, we need to go beyond Cmax and AUC and consider tumor tissue penetration and tumor saturation to design next-generation ADCs.
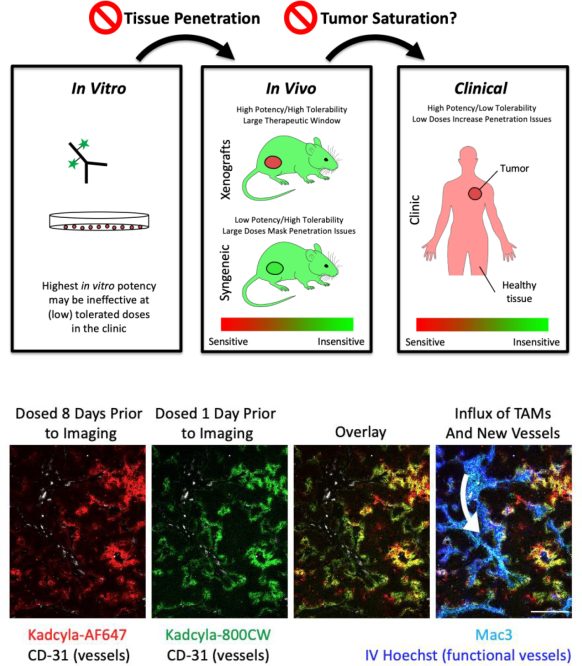
Tumor permeability
Systemic pharmacokinetics, toxicity, and efficacy are key metrics measured during drug development. Toxicity and pharmacokinetics can quickly screen out poor drug candidates at an early stage. In contrast, testing efficacy requires greater effort, and tissue penetration plays an important role in this process.
Tumor tissue permeability, which affects efficacy, cannot be considered alone, and toxicity is equally important. For example, at the clinical dose of 3.6 mg/kg, 43.1% of Kadcyla-treated patients showed ≥ grade 3 adverse reactions. Investigators are investigating alternative delivery methods using ADCs to potentially increase tolerability, such as batch dosing. However, when single-dose versus batch-dose Kadcyla efficacy was examined, the clinical benefit of batch-dose was reduced. Therefore, reduced dose and increased frequency of administration may be more tolerable, but smaller doses generally result in lower plasma concentrations, reduced tumor penetration, and lower tumor cell targeting.
Tumor saturation
Another critical aspect is tumor saturation. This depends on a variety of conditions, including dose (Cmax), expression (receptor/cell), rate of internalization and plasma clearance, etc. The importance of tumor saturation is twofold: saturating doses are more likely to be used in preclinical models; and saturating and subsaturating doses may have opposite results. First, the doses administered to mice did not always correspond to clinically tolerated doses. Doses are sometimes increased in response to faster clearance or less responsive tumor models in mice, or because they are better tolerated in mice. This may lead to saturation in preclinical models, while clinically tolerated doses may be subsaturated. Because mouse cells typically do not respond as well as human cells to ADC payloads, large doses are required, and these large doses may mask delivery problems in the clinic.
Second, preclinical findings at saturating doses may produce opposite results to those at subsaturating doses in the clinic. For example, if a saturating dose is given, increasing the DAR is more effective, while when a supersaturating dose is given, lowering the DAR may be more effective. Typically, doses are limited by payload toxicity, so comparisons are made at constant payload doses. When tumors are oversaturated, cancer cells receive the greatest amount of antibody, so having more payload per antibody will yield greater efficacy. For supersaturated doses, the opposite is true. In this case, the ADC cannot reach all cancer cells, and increasing the DAR (at a constant payload dose) will reduce the amount of antibody delivered, thereby reducing the number of cells targeted and killed. Conversely, lowering the DAR and/or increasing the total antibody dose under these conditions can improve tissue permeability and overall efficacy. Therefore, the payload MTD should be associated with a saturating dose of antibody for maximum tissue penetration and efficacy.
In addition, three recently approved ADCs for solid tumors use payloads that can have bystander effects. The bystander effect allows the payload to diffuse out of the target cell and into adjacent cells after release. In theory, the bystander payload could also improve tissue penetration beyond what the antibody itself could achieve. This may explain the increased efficacy exhibited by Enhertu compared to Kadcyla in the NCI-N87 mouse model, despite similar cellular potency. However, even when using ADCs with bystander payloads, the increased tissue penetration of higher antibody doses improved efficacy. Although bystander payload improved distribution, antibody direct delivery was more efficient than bystander killing, explaining that even with bystander payload, higher antibody doses had a greater effect.
summary
Target selection, linker stability, and payload toxicity have been major considerations in ADC drug design over the past few decades. Even so, many ADCs that looked promising in preclinical studies ended up failing in clinical trials due to toxicity and/or poor therapeutic windows.
By analyzing the shared features of recently FDA-approved ADC drugs, it can be found that tissue penetration and tumor saturation are also key to the successful design of ADCs. Therefore, data on the most important parameters of efficacy, including tumor penetration and saturation, require special attention in preclinical studies. In an ideal design, at least two pieces of information are known: the payload dose tolerable in humans and the antibody dose required to saturate the tumor target. Armed with this information, we can maximise efficacy at clinically tolerated doses by modifying the DAR to deliver the highest tolerated dose of payload to all tumor cells.
references:
1. Key metrics to expanding the pipeline of successful antibody-drug conjugates. Trends Pharmacol Sci. 2021 Oct; 42(10): 803–812.